The Fries rearrangement proceeds through ionic intermediates. The reaction depends on the structure of the substrates and the reaction conditions.
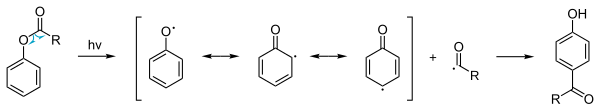
The scheme depicts the formation of an ortho-acylated phenol from a substituted phenolic ester in the presence of aluminium trihalide catalyst. The photo Fries rearrangement mechanism proceeds through Radical intermediates.
The Fries rearrangement proceeds through ionic intermediates. The reaction depends on the structure of the substrates and the reaction conditions.
The scheme depicts the formation of an ortho-acylated phenol from a substituted phenolic ester in the presence of aluminium trihalide catalyst. The photo Fries rearrangement mechanism proceeds through Radical intermediates.
The
Fries rearrangement, named for the German chemist
Karl Theophil Fries, is a
rearrangement reaction of a
phenyl ester to a
hydroxy aryl ketone by
catalysis of
Lewis acids.
[1][2][3][4]
It involves migration of an
acyl group of
phenyl ester to
benzene ring. The reaction is
ortho and para selective and one of the two products can be favoured by changing reaction conditions, such as
temperature and
solvent.
Mechanism
Despite many efforts a definitive
reaction mechanism for the Fries rearrangement is not available. Evidence for inter- and
intramolecular mechanisms have been obtained by so-called cross-experiments with mixed reactants. Reaction progress is not dependent on
solvent or
substrate. A widely accepted mechanism involves a
carbocation intermediate.
In the first reaction step a
Lewis acid for instance
aluminium chloride AlCl
3 co-ordinates to the
carbonyl oxygen atom of the
acyl group. This oxygen atom is more
electron rich than the
phenolic oxygen atom and is the preferred
Lewis base. This interaction
polarizes the
bond
between the acyl residue and the phenolic oxygen atom and the aluminium
chloride group rearranges to the phenolic oxygen atom. This generates a
free
acylium carbocation which reacts in a classical
electrophilic aromatic substitution with the aromatic ring. The abstracted proton is released as
hydrochloric acid
where the chlorine is derived from aluminium chloride. The orientation
of the substitution reaction is temperature dependent. A low reaction
temperature favors
para substitution and with high temperatures the
ortho
product prevails. Formation of the ortho product is also favoured in
non-polar solvents; as the solvent polarity increases, the ratio of the
para product also increases.
[5]
Scope
Phenols react to
esters but do not react to hydroxyarylketones with acylhalogen compounds under
Friedel-Crafts acylation
reaction conditions and therefore this reaction is of industrial
importance for the synthesis of hydroxyarylketones which are important
intermediates for several pharmaceutics such as
paracetamol and
salbutamol. As an alternative to
aluminium chloride, other
Lewis acids such as
boron trifluoride and
bismuth triflate or strong protic acids such as
hydrogen fluoride and
methanesulfonic acid can also be used. In order to avoid the use of these corrosive and environmentally unfriendly
catalysts altogether research into alternative
heterogeneous catalysts is actively pursued.
Limits
In all instances only
esters
can be used with stable acyl components that can withstand the harsh
conditions of the Fries rearrangement. If the aromatic or the acyl
component is heavily substituted then the
chemical yield will drop due to
steric constraints. Deactivating meta-directing groups on the benzene group will also have an adverse effect as can be expected for a
Friedel–Crafts acylation.
Photo-Fries rearrangement
In addition to the ordinary thermal phenyl ester reaction a so-called
photochemical Photo-Fries rearrangement exists
[6] that involves a
radical reaction mechanism. This reaction is also possible with deactivating
substituents
on the aromatic group. Because the yields are low this procedure is not
used in commercial production. However, photo-Fries rearrangement may
occur naturally, for example when a plastic bottle made of polyethylene
terephthalate (PET) is exposed to the sun, particular to UV light at a
wavelength of about 310 nm, if the plastic has been heated to 40 degrees
Celsius or above (as might occur in a car with windows closed on a hot
summer day). In this case, photolysis of the ester groups would lead to
leaching of phthalate from the plastic.
[7]
Anionic Fries rearrangment
In addition to Lewis acid and photo-catalysed Fries rearrangements,
there also exists an anionic Fries rearrangement. In this reaction, the
aryl ester undergoes ortho-metallation with a strong base, which then
rearranges in a nucleophilic attack mechanism.
- Fries, K. ; Finck, G. (1908). "Über Homologe des Cumaranons und ihre Abkömmlinge". Chemische Berichte 41 (3): 4271–4284. doi:10.1002/cber.190804103146.
- Fries, K.; Pfaffendorf, W. (1910). "Über ein Kondensationsprodukt des Cumaranons und seine Umwandlung in Oxindirubin". Chemische Berichte 43 (1): 212–219. doi:10.1002/cber.19100430131.
- March, J. Advanced Organic Chemistry, 3rd Ed.; John Wiley & Sons: Chichester, 1985; S. 499ff.
- Blatt, A. H. Org. React. 1942, 1.
- Kürti, László; Czakó, Barbara (2005). Strategic Applications of Named Reactions in Organic Synthesis: Background and Detailed Mechanisms. Elsevier Academic Press. p. 181. ISBN 0123694833.
- Bellus, D. Advances in Photochemistry; John Wiley & Sons: Chichester, 1971; Vol. 8, 109–159.
- Norma Searle, "Environmental effects on polymeric materials," pp. 313–358, in Plastics and the Environment, edited by Anthony Andrade, Wiley, 2003.