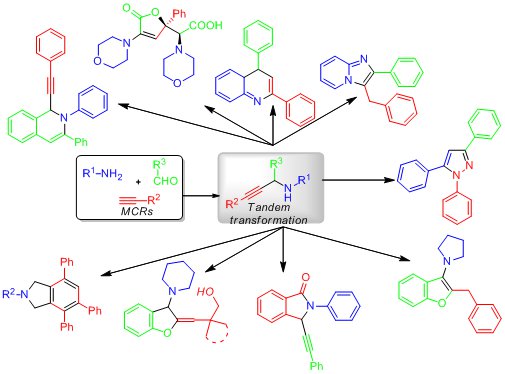
 Recent advances on diversity oriented heterocycle synthesis via multicomponent tandem reactions based on A3 coupling (14-8183LR) [pp. 1-20]
Arkivoc 2014 Part (i), 1-20: Special Issue 'Reviews and Accounts'Yunyun LiuFull Text: PDF (235K)
http://www.arkat-usa.org/get-file/48824/
Recent advances on diversity oriented heterocycle synthesis via multicomponent tandem reactions based on A3 coupling (14-8183LR) [pp. 1-20]
Arkivoc 2014 Part (i), 1-20: Special Issue 'Reviews and Accounts'Yunyun LiuFull Text: PDF (235K)
http://www.arkat-usa.org/get-file/48824/
Recent advances on diversity oriented heterocycle synthesis via
multicomponent tandem reactions based on A3 coupling
Yunyun Liu*
a,b
a Key Laboratory of Functional Small Organic Molecule, Ministry of Education,
Jiangxi Normal University, Nanchang 330022, P. R. China
b College of Chemistry and Chemical Engineering, Jiangxi Normal University,
Nanchang 330022, P. R. China
Abstract
A3 coupling reactions are the reactions between aldehydes, amines and alkynes, which yield
propargylamine derivatives under various catalyst conditions. By making use of the versatile
reactivity of propargylamines, tandem reactions initiated by the functional group(s) in the in situ
generated propargylamines constitute one of the most important applications of A3
couplings.
These tandem reactions are especially useful for the synthesis of heterocyclic compounds. In this
review, the progress on multicomponent tandem reactions based on A3
coupling is summarized.
Author’s Biography
Dr. Yunyun Liu was born in 1983 in Shandong Province, China. She obtained her Bachelor
Degree in Qufu Normal University in 2005. She then moved to Zhejiang University to continue
her graduate study in the Department of Chemistry. Under the supervision of Professor Weiliang
Bao, she worked on the field of copper-catalyzed Ullmann coupling reaction and related tandem
reactions for her doctorate study. She obtained her doctorate degree in 2010 and presently she is
an assistant professor in Jiangxi Normal University. She is currently interested in the research of
metal-catalyzed organic synthesis and the development of new cascade organic reactions.


 Open Access
Open Access
 Inexpensive hydrochloric acid is promising for low-cost
electrochemical chlorination
Inexpensive hydrochloric acid is promising for low-cost
electrochemical chlorination Palladium-catalyzed carbonylative α-arylation of aryl bromides
Palladium-catalyzed carbonylative α-arylation of aryl bromides The iodoaldol reaction of internal alkynyl ketones gives
useful oxygen-functionalized vinyl iodides
The iodoaldol reaction of internal alkynyl ketones gives
useful oxygen-functionalized vinyl iodides










 Production of chiral carboxylic esters proves a useful method
in the total synthesis of centrolobine
Production of chiral carboxylic esters proves a useful method
in the total synthesis of centrolobine