Asymmetric synthesis of ent-fragransin C1
Org. Biomol. Chem., 2017, Advance Article
DOI: 10.1039/C7OB00749C, Paper
DOI: 10.1039/C7OB00749C, Paper
Santikorn Chaimanee, Manat Pohmakotr, Chutima Kuhakarn, Vichai Reutrakul, Darunee Soorukram
The first asymmetric synthesis of ent-fragransin C1 bearing 2,3-anti-3,4-syn-4,5-anti stereochemistries is reported.
The first asymmetric synthesis of ent-fragransin C1 bearing 2,3-anti-3,4-syn-4,5-anti stereochemistries is reported.
Asymmetric synthesis of ent-fragransin C1

Abstract
The first asymmetric synthesis of ent-fragransin C1 was reported. The key step involves an intramolecular C–O bond formation (furan ring formation) via chemoselective generation of the benzylic carbocation leading to the 2,3-anti-3,4-syn-4,5-anti-tetrahydrofuran moiety as a single diastereomer in good yield. Our synthesis confirms that ent-fragransin C1 possesses 2R,3R,4S,5S configurations.
4-[(2R,3R,4S,5S)-5-(4-Hydroxy-3-methoxyphenyl)-3,4- dimethyltetrahydrofuran-2-yl]-2,6-dimethoxyphenol (ent-1). A flame-dried.......................... in vacuo, the crude product was purified by column chromatography (60% EtOAc in hexanes) to afford ent-1 as a sticky brownish oil (15.3 mg, 99% yield) as a single diastereomer (400 MHz 1 H NMR analysis). Rf 0.18 (40% EtOAc in hexanes);
[α]27 D -6.97 (c 0.60, CHCl3 ) (lit. [α]D +3.8 (c 0.60, CHCl3 ); 4a
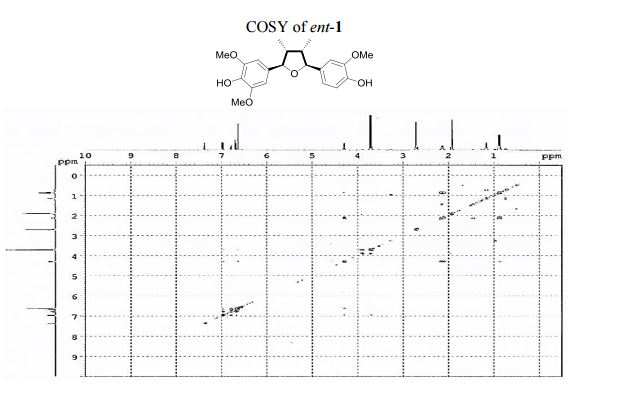
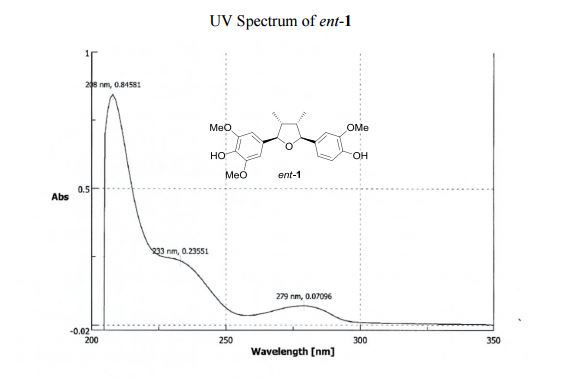
UV (MeOH) λmax (log ε) 208 (0.85), 233 (0.24), 279 (0.07) nm; CD (MeOH) 226 (Δε 2.23), 250 (Δε +0.12).
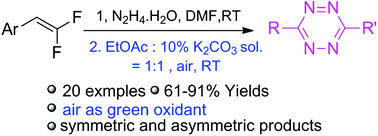
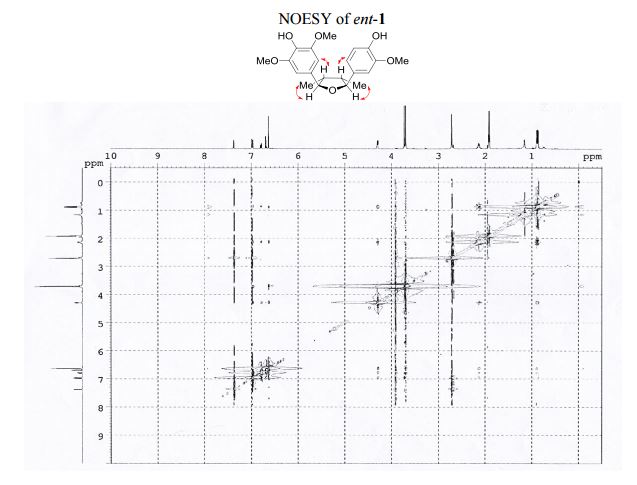
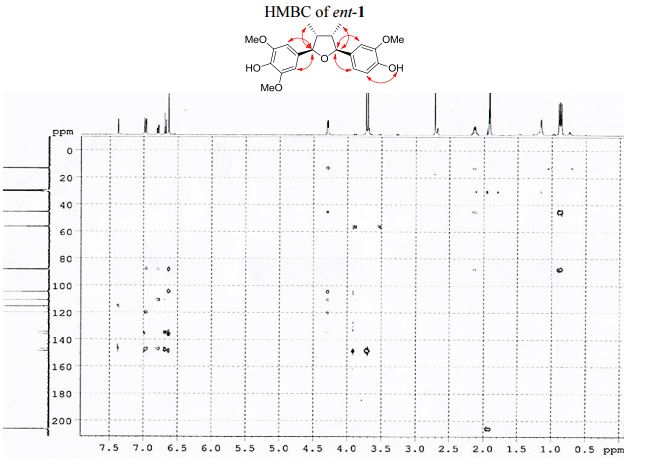
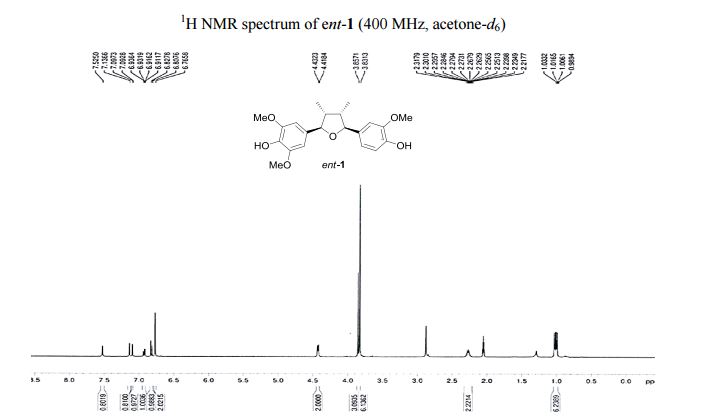
1 H NMR (400 MHz, acetone-d6 ): δ 7.51 (s, 1H, OH), 7.12 (s, 1H, OH), 7.10 (d, J = 1.8 Hz, 1H, ArH), 6.92 (d, J = 8.1, 1.8 Hz, 1H, ArH), 6.82 (d, J = 8.1 Hz, 1H, ArH), 6.77 (s, 2H, 2 ArH), 4.43 (d, J = 5.4 Hz, 1H, 2 xCH), 3.86 (s, 3H, OCH3 ), 3.83 (s, 6H, 2 xOCH3 ), 2.202.45 (m, 2H, 2 xCH), 1.03 (d, J = 6.7 Hz, 3H, CH3 ), 1.00 (d, J = 6.7 Hz, 3H, CH3 ).
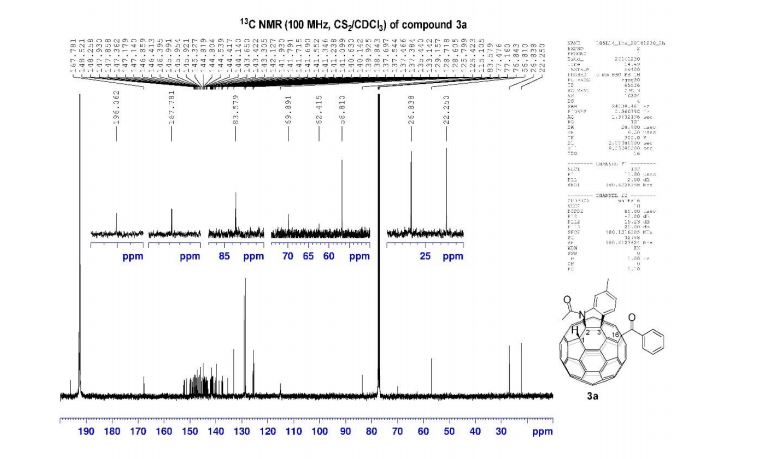
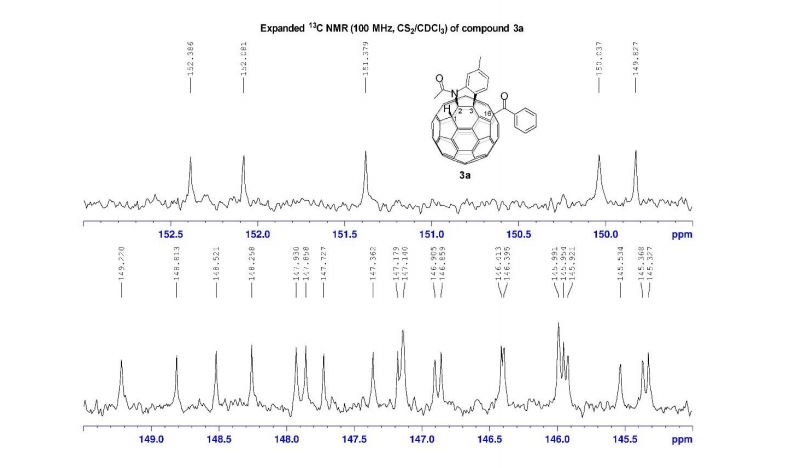
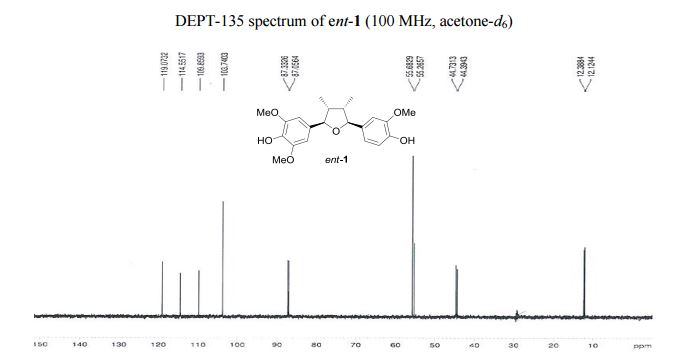
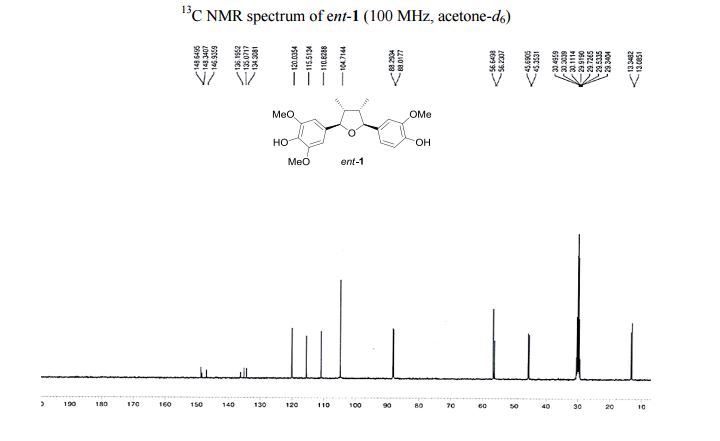
13C NMR (100 MHz, acetone-d6 ): δ 149.0 (2 C), 148.7 (C), 147.3 (C), 136.6 (C), 135.5 (C), 134.7 (C), 120.4 (CH), 115.9 (CH), 111.2 (CH), 105.1 (2 CH), 88.7 (CH), 88.4 (CH), 57.0 (2 CH3 ), 56.6 (CH3 ), 46.1 (CH), 45.7 (CH), 13.7 (CH3 ), 13.5 (CH3 ).
IR (CHCl3 ): νmax 3542s, 1616m, 1517s, 1465s, 1117s cm−1 .
MS (ISCID): m/z (%) relative intensity 397 [(M + Na)+ , 100], 357 (1).
HRMS (ESI-TOF) calcd for C21H26O6Na [M + Na]+ : 397.1627, found: 397.1622.
4 (a) M. Hattori, S. Hada, Y. Kawata, Y. Tezuka, T. Kikuchi and T. Namba, Chem. Pharm. Bull., 1987, 35, 3315; (


Darunee Soorukram
Assistant Professor
Darunee Soorukram
Education
- B.Sc. (Chemistry), Khon Kean University, Khon Kean, Thailand
- M.Sc. (Organic Chemistry), Mahidol University, Bangkok, Thailand
- Ph.D., Ludwig-Maximilians-University, Munich, Germany
Contact
Phone: +66.(0).2.201.5148
Address:
Room C418B
Department of Chemistry,
Faculty of Science, Mahidol University,
Rama 6 Road, Ratchthewee
Bangkok 10400 Thailand
Room C418B
Department of Chemistry,
Faculty of Science, Mahidol University,
Rama 6 Road, Ratchthewee
Bangkok 10400 Thailand
/////////