
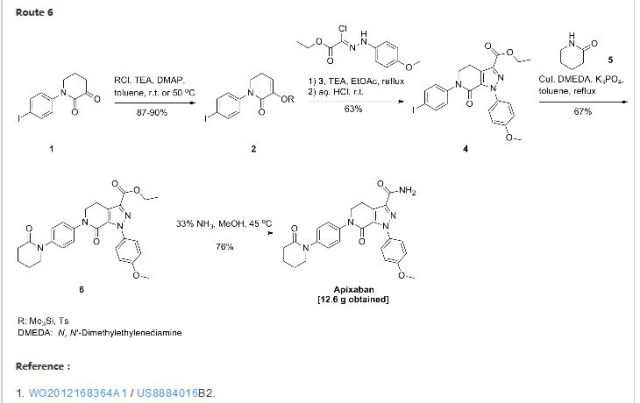
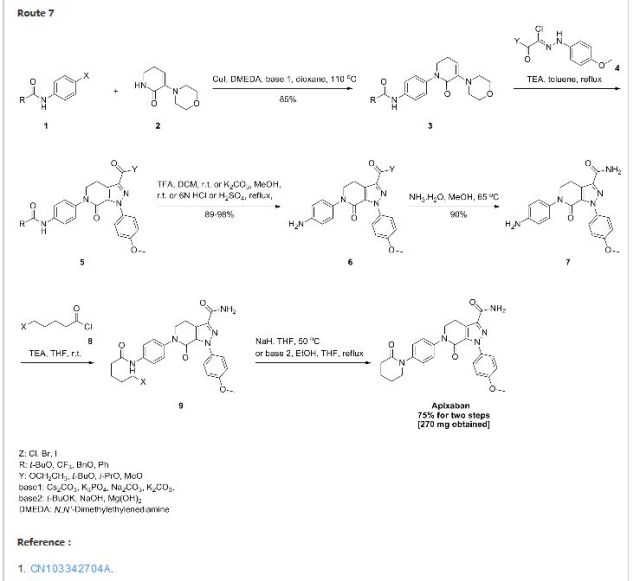
1 ZIFAXABAN
2 RIVAROXABAN
3 BETRIXABAN
4 EDOXABAN
5 APIXABAN
6
7
8
will be updated...........Xabans
8
will be updated...........Xabans
- Apixaban
- Betrixaban§
- Darexaban§
- Edoxaban
- Otamixaban§
- Rivaroxaban etc watch out
1
ZIFAXABAN
Zifaxaban



Zifaxaban
cas 1378266-98-8
rotation (-)
C20 H16 Cl N3 O4 S
C20H16ClN3O4 S, M = 429.87
Tianjin Institute of Pharmaceutical Research
Deep vein thrombosis; Lung embolism
Factor Xa antagonist
TY-602; zhifeishaban; zifaxaban
Chinese J Struc Chem. 2014, 33 (7), 1091-1095.
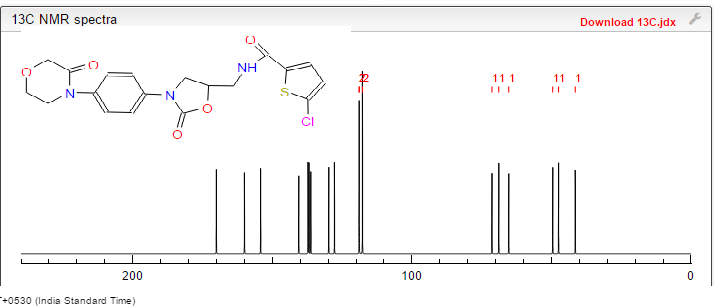
(S) -5- chloro -N- ((2- oxo _3_ (4_ (2_ oxo _2H_-1-yl) phenyl) oxazolidin-5 -1,3_ yl) methyl) thiophene-2-carboxamide
5-Chloro-N-(5S)-2-oxo-3-[4-(2-oxopyridin-1(2H)-yl)phenyl]oxazolidin-5-ylimethyllthiophene-2-carboxamide]
The title compound(zifaxaban 2, C20H16ClN3O4 S, Mr = 429.87) was synthesized and its crystal structure was determined by single-crystal X-ray diffraction. Zifaxaban crystallizes in monoclinic, space group P21 with a = 5.7900(12), b = 13.086(3), c = 12.889(3) A, β = 100.86(3)°, V = 959.1(3) A3, Z = 2, Dc = 1.489 g/cm3, F(000) = 444, μ = 0.342 mm-1, the final R = 0.0320 and wR = 0.0640 for 2717 observed reflections(I > 2σ(I)). The absolute configuration of the stereogenic center in the title compound was confirmed to be S by single-crystal X-ray diffraction. Four existing intermolecular hydrogen bonds help to stabilize the lattice and the molecule in the lattice to adopt an L-shape conformation. Zifaxaban was slightly more active than rivaroxaban 1 in in vitro assay against human FXa and therefore is promising as a drug candidate.
zifaxaban (first disclosed in CN102464658), useful for treating thromboembolic disorders. Zifaxaban, a factor Xa antagonist, is being developed by Tianjin Institute of Pharmaceutical Research, for treating deep vein thrombosis and pulmonary embolism (preclinical, as of November 2014). In May 2014, an IND was filed in China. In June 2014, the institute was seeking to outlicense this product.
In vivo within the cardiovascular, blood coagulation or blood analysis some have formed out of the process of forming a solid mass with the aggregation, called thrombosis, the formation of a solid mass called a thrombus blocks.Thrombosis is an abnormal flow of blood coagulation status due to platelet activation and coagulation factors are activated in accordance therewith.
The blood coagulation was originally a protective mechanism of the organism, there is a mutual antagonism in blood coagulation system and the anti-clotting system. Under physiological conditions, blood clotting factors continue to be activated to produce thrombin, fibrin formation trace, calm on the vascular endothelium, but these traces of fibrin and constantly being activated fibrinolytic system dissolution, while being activated coagulation factors are constantly mononuclear phagocyte system swallowed. The dynamics of the coagulation system and fibrinolysis system, which ensures the blood coagulation potential can also always ensure that the fluid state of the blood.
Sometimes, however, in certain factors can promote the coagulation process, breaking the above dynamic balance triggered the coagulation process, the blood can form a thrombosis or embolism, such as leading to myocardial infarction, stroke, deep vein thrombosis, pulmonary embolism and other thromboembolic disease. Thromboembolic disease is cardiovascular disease against the most serious diseases, is the first killer of human health. In China, with the improvement and increased aging of the population’s living standards, the incidence of such diseases, mortality, morbidity is increasing every year.
The existing anti-thromboembolic diseases into anti-platelet drugs, anticoagulants and fibrinolytic drugs. Among them, the anti-clotting drugs are the main contents of antithrombotic therapy, mainly thrombin inhibitors and vitamin K antagonists. Heparin and low molecular weight heparin, represented by the presence of oral thrombin inhibitor invalid, non-selective inhibition and high risk of bleeding and other shortcomings. Although warfarin is representative of vitamin K antagonists can be administered orally, but there are narrow therapeutic index, high risk of bleeding and other shortcomings.
Studies have shown that the coagulation process is usually divided into intrinsic coagulation pathway and the extrinsic coagulation pathway. Coagulation process involves a lot of coagulation factors, coagulation factor activated are each the next inactive clotting factor precursor is converted into the activated form. Endogenous, exogenous pathway final summary, the blood coagulation factor X is converted to Xa. Therefore, theoretically, the direct inhibition of ¾ factor activity should produce effective anti-clotting effect, without the side effects of thrombin inhibitors with. As direct inhibition) (a factor activity on normal hemostasis reaction / adjustment process produces minimal impact. For example, platelets remain low catalytic activity of thrombin on the ability to respond to, and thus does not affect the formation of platelet thrombi, so bleeding integrated minimize the risk of the levy.
research also proved this point. Recently reported a variety of compounds can selectively inhibit efficient Xa, which play a preventive and / or treatment of thromboembolic disease effect (W003000256A1; CN00818966; US2007259913A1; US2007259913A1). Among them, rivaroxaban (Rivaroxaban) was listed in 2008 for hip or knee replacement surgery prophylaxis and treatment of venous thrombosis, with oral, fixed dose and other advantages.
rivaroxaban drawback is the high price of raw materials, low yield preparation, purification of the product is difficult, high production costs. Patent CN00818966 8 reported rivaroxaban synthetic routes as follows:
4

where the first reaction (Preparation of 4- (4-morpholino-3-yl) nitrobenzene) yield of only 17.6%, and rivaroxaban difficult purification.

………………………………
(S) -5- chloro -N- ((2- oxo-3- (4- (2_ oxo -2H- pyridin-1-yl) phenyl) -1, 3_ oxazolidine -5 – yl) methyl) thiophene-2-carboxamide.
[0011] Meanwhile, patent CN201110337461.4 described formula (I) Preparation of the compound:
[0012]

……………………………………..
CN102464658
Example 1
[0046] (S) -5- chloro -N- ((2- oxo-3- (4_ (2_ Batch oxo _2H_ piperidinyl) phenyl) _1,3_ oxazolidin-5-yl) methyl ) thiophene-2-carboxamide (II)
[0048] A, 1- (4- amino-phenyl) -IH- pyridin _2_ -one (Compound VII) is
[0049] The reaction flask was charged with 104g of pyridine -2 (IH) – one (Compound IX), 200g of iodoaniline (compound VIII), 26gCuI, 151g of potassium carbonate, 18g8- hydroxyquinoline, 500mlDMF, nitrogen, heated to reflux, Insulation reaction was stirred 10h. Filtered hot, the filtrate evaporated under reduced pressure to make the solvent, the residue was added ethyl acetate, IL, 0 ° C incubated with stirring lh, filtered and the solid dried, 2L acetonitrile and purified to give 98g dark red solid. Refined liquor was concentrated to 500ml, the ice bath was stirred lh, filtered to give a dark red solid 19g. Total product were 117g, yield 68.9%.
[0050] 1H-NMR (DMSO-Cl6), δ (ppm):… 5 306 (s, 2H), 6 236 (d, 1H), 6 406 (d, 1H), 6 601 (d,. 2H), 6. 977 (d, 2H), 7. 459 (m, 2H).
[0051] B, (R) -2- (2- hydroxy-3- ((2-oxo–2H- pyridin-1-yl) phenyl) amino) propyl) isoindoline-1,3- -dione (Compound V) is
[0052] The reaction flask was added 40gl_ (4- aminophenyl) -IH- pyridin-2-one (Compound VII), 45g (S) _N_ glycidyl phthalimide (Compound VI), 300ml95% ethanol, heating to reflux, the gradual emergence of solid insulation mixing IOh, cooled to room temperature, filtered, and the filter cake washed with ethanol (150ml X 2), and dried to give an off-white solid 38g.
[0053] The mother liquor was taken, evaporated to dryness under reduced pressure, was added 15g (Q-N_ glycidyl phthalimide (Compound VII), 150ml95% ethanol, heated to reflux, stirred incubated 10h, concentrated under reduced pressure, cooled to room temperature , stirred at room temperature for 2h, washed with ethanol and dried to give an off-white solid 33g.
[0054] A total of an off-white solid 71g, yield of 84.8%, without purification, was used directly in the next step.
[0055] 1H-NMR (DMS0_d6), δ (ppm):… 3 053 (m, 1H), 3 194 (m, 1H), 4 644 (m, 2H), 4 020 (m, 1H). , 5. 168 (d, 1H), 5. 851 (t, 1H), 6. 230 (m, 1H), 6. 404 (d, 1H), 6. 665 (d, 2H), 7. 041 ( d, 2H), 7. 435 (m, 1H), 7. 537 (m, 1H), 7. 855 (m, 4H).
[0056] C, ⑶-2- ((2- oxo-3- (4- (2_ oxo _2H_ pyridyl) phenyl) oxazolidin _5_ -1,3_ yl) methyl ) Preparation of isoindoline-1,3-dione (Compound IV) of the
[0057] The reaction flask was charged 50g Compound V, 27gN, N’- carbonyldiimidazole (⑶I), 4_ catalytic amount of dimethylaminopyridine (DMAP), 150mlN, N- dimethylformamide (DMF), stirred for 90 temperature ° C, the reaction was kept for 8 hours to make the solvent was evaporated under reduced pressure, added to IL of water, stirred and dispersed, filtered, washed with water (150mlX “, washed with ethanol (100ml X 1), dried to give a white solid 48g, yield of 90%.
[0058] 1H-NMR (DMSo-CI6), δ (ppm):…. 3 984 (m, 3H), 4 251 (t, 1H), 4 968 (m, 1H), 6 301 (m, 1H), 6. 459 (d, 1H), 7. 423 (d, 2H), 7. 514 (m, 1H), 7. 615 (m, 3H), 7. 892 (m, 4H).
[0059] D, (S) -5- (aminomethyl) -3- (4- (2_ oxo _2H_-1-yl) phenyl) oxazolidin _2_ -1,3_ one hydrochloride (compound III) Synthesis of
[0060] The reaction flask was charged 50g compound IV, 200ml of ethanol, 60ml aqueous methylamine (40%), heated to reflux, stirred incubated 2h, cooled, evaporated under reduced pressure to make the solvent to give a sticky solid.
[0061] added to 300ml of ethanol, 20ml of hydrochloric acid, heated to reflux, stirred incubated lh, cooled to room temperature, incubated with stirring 2h, filtered, washed with ethanol, and dried to obtain;. 34 5g of white solid, yield 88.7%.
[0062] 1H-NMR (DMS0_d6), δ (ppm):…. 3 240 (m, 2H), 3 980 (m, 1H), 4 255 (m, 1H), 5 028 (m, 1H) , 6. 321 (m, 1H), 6. 475 (d, 1H), 7. 504 (m, 3H), 7. 634 (m, 3H), 8. 561 (s, 1H).
[0063] Ε, (S) -5- chloro -N – ((2- oxo-3- (4- (2-oxo–2Η- pyridin-1-yl) phenyl) oxazolidin _1,3_ 5-yl) methyl) thiophene-2-carboxamide Preparation of thiophene (II) of
[0064] The reaction flask was charged 15g Compound III, 200ml of tetrahydrofuran, 40ml of water was added with stirring 6. 2g of sodium carbonate was added dropwise 10g5- chloro-thiophene-2-carbonyl chloride (Compound II-1) in tetrahydrofuran IOOml, 30~35 ° C insulation stirred 5h, point board to control the reaction was complete.
[0065] to make the solvent was distilled off under reduced pressure, 50ml of water was added, stirring was filtered, the filter cake washed with water and dried to give 18. 5g of white solid.
[0066] 200ml of acetic acid and purified room temperature overnight, filtered, and the filter cake washed with ethanol and dried to give a white solid 16g, 80% yield.
. [0067] Melting point: 204 8 ~205 8 ° C;
[0068] 1H-NMR (DMSo-CI6), δ (ppm):…. 3 623 (t, 2H), 3 893 (m, 1H), 4 230 (t, 1H), 4 871 (m, 1H), 6. 308 (t, 1H), 6. 468 (d, 1H), 7. 193 (d, 1H), 7. 426 (m, 2H), 7. 500 (m, 1H), 7. 637 (m, 4H), 8. 967 (t, 1H);
[0069] MS (ESI): m / z = 430 (M + H);
[0070] HPLC: rt (%) = 14. 38 (99. 62);
. [0071] [a] 20d = -37 6 ° (c 0. 3004, DMS0);
WO-2014183667Acetic acid solvate of oxazolidinone derivative, preparation method for the solvate, and application thereof
WO-2014183665Oxazolidinone derivative crystal form I and preparation method and use thereof
WO-2014183666Oxazolidinone derivate crystal form II, preparation method therefor, and application thereo
2 RIVAROXABAN


STRUCTURE
SIMILARITY
Chemical structures of linezolid (top) and rivaroxaban (bottom). The shared structure is shown in blue.







Wockhardt Ltd
The synthesis of (II) via intermediate (I) is described (example 7, page 15)
4-{4-[(5S)-5-(Aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one (formula III) is (I) and rivaroxaban is (II) (claim 1, page 16).
The present invention relates to a process for the preparation of Rivaroxaban and its novel intermediates, or pharmaceutically acceptable salts thereof. The present invention provides novel intermediates, which may be useful for the preparation of Rivaroxaban or its pharmaceutically acceptable salts thereof. The process of preparation by using novel intermediate is very simple cost effective and may be employed at commercial scale. The product obtained by using novel intermediate yield the Rivaroxaban of purity 99% or more, when measured by HPLC. The present invention especially relates to a process for the preparation of Rivaroxaban from thioester of formula II, or a pharmaceutically acceptable salt thereof, wherein R is leaving group.
process includes the step of , reacting thioester of formula IIA or pharmaceutically acceptable salt thereof
Formula IIA


with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one of formula III,
Formula III

Formula I

EXAMPLE 7: One pot process for Rivaroxaban
The triphenylphosphine (11.5g) and mercaptobenzothiazole disulphide (15.31g) were taken in methylene chloride and reaction mixture was stirred at 28°C -30°C for 1 hr. The 5-chlorothiophene-2-carboxylic acid (7.2g) and triethylamine (3.8 g) were added to the above reaction mixture. The reaction mixture is stirred at 0°C -25 °C for 1 hr. after 1 hr 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one (lOg) and triethylamine (3.8g) were added. The resulting reaction mixture further stirred for 2 hrs. After completion of the reaction, water was added and stirred for 10 min. aqueous layer was separated and washed with methylene chloride. The organic layer was acidified to pH 6-7 with 2N hydrochloric acid and finally the organic layer was concentrated to get desired product. The product was purified and dried to yield Rivaroxaban.
Yield: 10.0 gm
Purity: 99.3 %
EXAMPLE 8: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent ethyl acetate and base potassium hydroxide were used to get the rivaroxaban.
EXAMPLE 9: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent acetonitile and base potassium carbonate were used, methylene chloride was added in the reaction mixture to extract the Rivaroxaban.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015104605&recNum=7&maxRec=57790&office=&prevFilter=%26fq%3DOF%3AWO%26fq%3DICF_M%3A%22C07D%22&sortOption=Pub+Date+Desc&queryString=&tab=PCTDescription












Preparation 6 rivaroxaban implementation
 The
12.5 g (76.9 mmol) 5- chloro-thiophene-2-carboxylic acid was suspended
in 35 g of toluene was heated to 80 で, at this temperature, a solution
of 11.0 g (92.5 mmol) of thionyl chloride, reaction was continued for 30
min; then warmed to the boiling point of toluene was 120 ° C, and
stirring was continued under reflux until cessation of gas; cooled to
room temperature, the reaction mixture was concentrated under reduced
pressure to remove excess thionyl chloride and toluene to give
5-chloro-thiophene-2-carbonyl chloride;
The
12.5 g (76.9 mmol) 5- chloro-thiophene-2-carboxylic acid was suspended
in 35 g of toluene was heated to 80 で, at this temperature, a solution
of 11.0 g (92.5 mmol) of thionyl chloride, reaction was continued for 30
min; then warmed to the boiling point of toluene was 120 ° C, and
stirring was continued under reflux until cessation of gas; cooled to
room temperature, the reaction mixture was concentrated under reduced
pressure to remove excess thionyl chloride and toluene to give
5-chloro-thiophene-2-carbonyl chloride;
The 11.6 g (37.0 mmol) 4- {4 - [(5S) -5- (aminomethyl) -2-oxo-1,3-oxazolidin-3-yl] phenyl} morpholin-3 -one hydrochloride was added 40ml of water, was added 4. 64 g (43 8 mmol.) Na2CO3 stirred and dissolved; then added 50 ml of toluene, was added dropwise at 10 ° C under the mixture, the mixture is 8. 0 g ( 44. 4 mmol) 5- chloro-thiophene-2-carbonyl chloride was dissolved in 15 ml of toluene, 20 min the addition was complete, then stirring was continued at room temperature, TLC monitoring progress of the reaction, 2 h after completion of the reaction; and the filter cake washed with water and washed with acetone to give a pale yellow solid 19. 6 g, used directly ko acid recrystallization, as a white solid 15. 2 g,
mp 227. 2 - 228. 1 ° C, [a] D21 = -38 2 ° (. c = 0. 30, DMS0), rivaroxaban yield of 94%, the total yield of 87.5% 0
1H-NMR (DMSO) 8: 3. 61 (. 2 H, t, / = 5 4 Hz), 3. 71 (2 H, t, / = 5 4 Hz.), 3.85 (IH, m ), 3.97 (2 H, t, J = 4. 5 Hz), 4. 19 (3 H, t, / = 7. 5 Hz), 4.84 (IH, m), 7. 19 (IH, d, / = 4. 2Hz), 7.40 (2 H, d, /=9.0 Hz), 7. 57 (2 H, t, /=9.0 Hz), 7. 69 (IH, d, J = 4. 19 Hz), 8. 96 (IH, t, / = 5. 7 Hz).
 10
g of the salt prepared according to Example 18 were suspended in 75 ml
of N- methylpyrolidone, the suspension was heated at 50°C, then 14 ml of
triethylamine was added and the mixture was heated at 60°C. This was
followed by addition of 15.7 ml of a solution of
5-chlorothiophene-2-carboxylic acid chloride in toluene (2.46 M) and the
reaction mixture was stirred and heated at 55°C for 15 minutes, then
slowly cooled below 30°C, 75 ml were added and the turbid solution was
filtered. The clear filtrate was stirred at 50°C, which was followed by
addition of 15 ml of water and 75 ml of ethanol and stirring for 1 hour
under slow cooling. The separated product was filtered off, washed with
water (15 ml, 60°C), ethanol (2 x 25 ml) and dried in vacuo. 9.1 g
(yield 81%) of rivaroxaban in the form of an off-white powder with the
melt, point of 229.5-231°C was obtained, HPLC 99.95%, content of the (
)-isomer below 0.03%.
10
g of the salt prepared according to Example 18 were suspended in 75 ml
of N- methylpyrolidone, the suspension was heated at 50°C, then 14 ml of
triethylamine was added and the mixture was heated at 60°C. This was
followed by addition of 15.7 ml of a solution of
5-chlorothiophene-2-carboxylic acid chloride in toluene (2.46 M) and the
reaction mixture was stirred and heated at 55°C for 15 minutes, then
slowly cooled below 30°C, 75 ml were added and the turbid solution was
filtered. The clear filtrate was stirred at 50°C, which was followed by
addition of 15 ml of water and 75 ml of ethanol and stirring for 1 hour
under slow cooling. The separated product was filtered off, washed with
water (15 ml, 60°C), ethanol (2 x 25 ml) and dried in vacuo. 9.1 g
(yield 81%) of rivaroxaban in the form of an off-white powder with the
melt, point of 229.5-231°C was obtained, HPLC 99.95%, content of the (
)-isomer below 0.03%.
1H NMR (250 MHz, DMSO-D6), δ (ppm): 3.61 (t, 2H, CH2); 3.71 (m, 2H, CH2); 3.85 and 4.19 (m, 2x1 H, CH2); 3.97 (m, 2H, CH2); 4.19 (s, 2H, CH2); 4.84 (pent, 1H, CH); 7.18 (d, 1H); 7.40 (m, 2H); 7.56 (m, 2H); 7.68 (d, 1H); 8.95 (bt, 1H, NH).
13C NMR (250 MHz, DMSO-D6), δ (ppm): 42.2; 47.4; 49.0; 63.4; 67.7; 71.3; 1 18.3; 125.9; 128.1 ; 128.4; 133.2; 136.4; 137.0; 138.4; 154.0; 160.8; 165.9.
MS (m/z): 436.0729 (M+H)+. ation)
 The
optical isomer of rivaroxaban with the (R)- configuration was obtained
by a process analogous to Example 28 starting from the salt prepared
according to Example 19. The yield was 76%, HPLC 99.90%, content of the
(5)-isomer below 0.03%. The NMR and MS spectra were in accordance with
Example 28.
The
optical isomer of rivaroxaban with the (R)- configuration was obtained
by a process analogous to Example 28 starting from the salt prepared
according to Example 19. The yield was 76%, HPLC 99.90%, content of the
(5)-isomer below 0.03%. The NMR and MS spectra were in accordance with
Example 28.
..........................

........................
5- chloro-thiophene-2-chloride by condensation, bromide, with 4- (4-amino-phenyl) -3-morpholinone cyclization reaction rivaroxaban, the following reaction scheme :( References : W02005068456, US20070149522, DE10300111)






http://www.google.com/patents/CN102702186A?cl=zh
Compound rivaroxaban Synthesis Example 7 formula (X), [0071] Example
[0072] Method One:
 [0074] The compound of formula (VIII) of (180mg, 0. 618mmol), Ni chloride (5mL) and tris ko amine (187mg,
[0074] The compound of formula (VIII) of (180mg, 0. 618mmol), Ni chloride (5mL) and tris ko amine (187mg,
I. 85mmol) added to the reaction flask, stirred at room temperature for 10 minutes, cooled to 0 ° C, a solution of 5-chloro-2-thiophene chloride (224mg, 1.24mm0l), stirred at room temperature overnight; after the completion of the reaction, spin dry, rinse with anhydrous alcohol ko, filtered, washed ko anhydrous alcohol three times to obtain a white solid product rivaroxaban (215mg, embodiments of the total yield of 7,8 80%).
[0075] 1H-Mffi (DMSC) JOOMHz, δ d m):.... 3 61 (t, 2H, J = 5 6Hz), 3. 71 (t, 2H, J = 5 2Hz), 3 89 ( m, 1H), 3. 97 (t, 2H, J = 4. 4Hz), 4. 20 (m, 3H), 4. 85 (m, 1H), 7. 18 (d, 1H, J = 4. 0Hz), 7. 40 (d, 2H, J = 8. 8Hz), 7. 56 (d, 2H, J = 8. 8Hz), 7. 73 (d, 1H, J = 4. 0Hz).
The method of writing is:
 [0078]
The compound 5_ gas - oh -I- thiophene carboxylic acid (500mg, 3.
08mmol), MsCl (702mg, 6. 1 Bmmol) and sodium bicarbonate (. 517mg, 6
16mmol) was suspended in THF (20ml) in , heated to 60 ° C with stirring
45min, a large white suspension washed out; the reaction mixture was
cooled to room temperature, the compound of formula VIII was added
portionwise (800mg, 2 75mmol.), stirred for 5 hours, after completion of
the reaction distilled THF, was added after the residue was cooled to
room temperature, water (IOOml), at room temperature embrace Cheung
30min, filtered, and the filter cake washed with cold water, dried and
added to a ko-ol (5ml) was heated at reflux for I hour. After cooling,
stirred for 5 hours at room temperature After filtration to give the
product of formula (X) of the compound rivaroxaban (719mg, 60%)
[0078]
The compound 5_ gas - oh -I- thiophene carboxylic acid (500mg, 3.
08mmol), MsCl (702mg, 6. 1 Bmmol) and sodium bicarbonate (. 517mg, 6
16mmol) was suspended in THF (20ml) in , heated to 60 ° C with stirring
45min, a large white suspension washed out; the reaction mixture was
cooled to room temperature, the compound of formula VIII was added
portionwise (800mg, 2 75mmol.), stirred for 5 hours, after completion of
the reaction distilled THF, was added after the residue was cooled to
room temperature, water (IOOml), at room temperature embrace Cheung
30min, filtered, and the filter cake washed with cold water, dried and
added to a ko-ol (5ml) was heated at reflux for I hour. After cooling,
stirred for 5 hours at room temperature After filtration to give the
product of formula (X) of the compound rivaroxaban (719mg, 60%)
SYN 1

BAYER HEALTHCARE AG Patent: WO2004/60887 A1, 2004 ; Location in patent: Page/Page column 8; 10-11 ;
SYN 2


MEDICHEM S.A.; MANGION, Bernardino; DURAN LOPEZ, Ernesto Patent: WO2012/35057 A2, 2012 ; Location in patent: Page/Page column 34 ;
SYN 3

EGIS GYOGYSZERGYAR NYILVANOSAN MUeKOeDOe RESZVENY-TARSASAG; SIPOS, Eva; KOVANYINE LAX, Gyoergyi; HAVASI, Balazs; VOLK, Balazs; KRASZNAI, Gyoergy; RUZSICS, Gyoergy; BARKOCZY, Jozsef; TOTHNE LAURITZ, Maria; LUKACS, Gyula; BOZA, Andras; HEGEDUeS, Laszlo Jozsef; TABORINE TOTH, Maria Julia; PECSI, Eva Patent: WO2012/153155 A1, 2012 ; Location in patent: Page/Page column 49 ;
SYN4
EGIS GYOGYSZERGYAR NYILVANOSAN MUeKOeDOe RESZVENY-TARSASAG; SIPOS, Eva; KOVANYINE LAX, Gyoergyi; HAVASI, Balazs; VOLK, Balazs; KRASZNAI, Gyoergy; RUZSICS, Gyoergy; BARKOCZY, Jozsef; TOTHNE LAURITZ, Maria; LUKACS, Gyula; BOZA, Andras; HEGEDUeS, Laszlo Jozsef; TABORINE TOTH, Maria Julia; PECSI, Eva Patent: WO2012/153155 A1, 2012 ; Location in patent: Page/Page column 68 ;
SYN 5
INTERQUIM, S.A.; Berzosa Rodríguez, Xavier; Marquillas Olondriz, Francisco; Llebaria Soldevilla, Amadeo; Serra Comas, Carme Patent: US2014/128601 A1, 2014 ; Location in patent: Paragraph 0068 ;
SYN 6
MEGAFINE PHARMA (P) LTD; MATHAD Vijayavitthal Thippannachar; PATIL NILESH SUDHIR, Nilesh; NIPHADE NAVNATH CHINTAMAN, Navnath; MALI ANIL CHATURLAL, Anil; BODAKE MAHENDRA BHAGIRATH, Mahendra; IPPAR SHARAD SUBHASH, Sharad; TALLA RAJESH, Rajesh Patent: WO2013/121436 A2, 2013 ; Location in patent: Page/Page column 31 ;
FROM THE NET
Rivaroxaban,
a FXa inhibitor, is the active ingredient in XARELTO Tablets with the
chemical name
5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-1,3-oxazolidin-5yl}methyl)-2-thiophenecarboxamide.
The molecular formula of rivaroxaban is C19H18ClN3O5S and the molecular weight is 435.89. The structural formula is:
Rivaroxaban
is a pure (S)-enantiomer. It is an odorless, non-hygroscopic, white to
yellowish powder. Rivaroxaban is only slightly soluble in organic
solvents (e.g., acetone, polyethylene glycol 400) and is practically insoluble in water and aqueous media.
Each XARELTO tablet contains 10 mg, 15 mg, or 20 mg of rivaroxaban. The inactive ingredients of XARELTO are: croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. Additionally, the proprietary film coating mixture used for XARELTO 10 mg tablets is Opadry® Pink and for XARELTO 15 mg tablets is Opadry® Red, both containing ferric oxide red, hypromellose, polyethylene glycol 3350, and titanium dioxide, and for XARELTO 20 mg tablets is Opadry® II Dark Red, containing ferric oxide red, polyethylene glycol 3350, polyvinyl alcohol (partially hydrolyzed), talc, and titanium dioxide.
3
BETRIXABAN
CAS 330942-05-7
MW 451.91, C23H22ClN5O3
Venous Thromboembolism (VTE)
Millennium INNOVATOR
Takeda Pharmaceutical Co Ltd
Lee’s Pharmaceutical Holdings (Hong Kong) Ltd; Portola Pharmaceuticals Inc…DEVELOPERS
PHASE 3 for Venous Thromboembolism (VTE)

Patents CN1391555A, CN102336702A, CN101595092A, CN102762538A
Portola Pharmaceuticals, under license from Takeda (formerly known as Millennium Pharmaceuticals), is developing betrixaban (was reported to be in phase III in November 2015), for treating venous thrombosis
In October 2015, betrixaban was granted Fast Track designation by the FDA for extended-duration prevention of VTE or blood clots in acute medically ill patients
Betrixaban (INN, codenamed PRT-054,021) is an anticoagulant drug which acts as a direct factor Xa inhibitor.[1] It is potent, orally active and highly selective for factor Xa, being selected from a group of similar compounds for its low hERG affinity.[2] Betrixaban has undergone human clinical trials for prevention of embolism after knee surgery,[3] and prevention of stroke following atrial fibrillation,[4] with promising results.[5] Betrixaban is currently being studied in a human clinical trial for extended duration thromboprophylaxis to prevent venous thromboembolism in acute medically ill patients.[6] Joint development with Portola was discontinued in 2011 by Merck.[7] Betrixaban is now being developed by Portola Pharmaceuticals.


Portola has initiated its pivotal Phase 3 APEX Study to demonstrate the safety and efficacy of betrixaban for extended duration venous thromboembolism (VTE) prophylaxis for up to 35 days in acute medically ill patients with restricted mobility and certain risk factors. This randomized, double-blind, active-controlled, multicenter, multinational study will compare a once-daily dose of 80 mg of betrixaban for a total of 35 days (including both in the hospital and after discharge) with in-hospital administration of 40 mg of enoxaparin once daily for 6 to 14 days followed by placebo. The global study is expected to enroll approximately 6,850 patients at more than 400 study sites throughout the world. The primary objective of the trial is to demonstrate the superiority of betrixaban as compared to the current standard of care in the reduction of VTE-related events at 35 days while maintaining a favorable benefit to risk profile.
The APEX study is adequately powered to show a clinically relevant benefit with a p-value of less than 0.01 on the primary endpoint of total asymptomatic proximal DVT (as detected by ultrasound), symptomatic DVT (proximal or distal), non-fatal pulmonary embolism and VTE-related death. The first patient was enrolled in March 2012.
The safety and tolerability of betrixaban for stroke prevention was evaluated in 508 patients with atrial fibrillation in the Phase 2 EXPLORE-Xa dose-ranging study. Results were presented during a late-breaking session at the American College of Cardiology’s 59th Annual Scientific Session in March 2010. The data showed that a once-daily dose of oral betrixaban, given to patients with non-valvular atrial fibrillation or atrial flutter and at least one risk factor for stroke, reduced the incidence of major and clinically relevant non-major bleeds compared to dose-adjusted warfarin. In August 2010, additional pharmacodynamic data from a pre-specified analysis of EXPLORE-Xa showed a concentration dependent relationship and provided further evidence for the anticoagulant activity of betrixaban across all three doses studied in the clinical trial. The additional pharmacodynamic analysis provides information for dose selection for Phase 3 evaluation of betrixaban.
In 2007, positive top-line results from EXPERT were published in The Journal of Thrombosis and Haemostasis. This randomized, multi-center, Phase 2 in-hospital efficacy and safety study of the prevention of VTE compared betrixaban with enoxaparin in 215 patients undergoing knee replacement surgery.

Betrixaban (INN, codenamed PRT-054,021) is an anticoagulant drug which acts as a direct factor Xa inhibitor.[1] It is potent, orally active and highly selective for factor Xa, being selected from a group of similar compounds for its low hERG affinity.[2] Betrixaban has undergone human clinical trials for prevention of embolism after knee surgery,[3] and prevention of stroke following atrial fibrillation,[4] with promising results.[5]
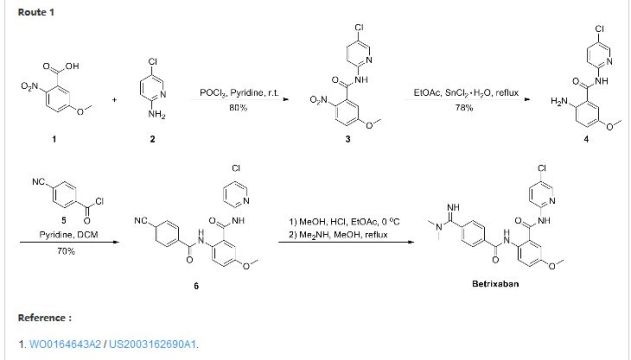

Patent Document CN1391555A first discloses a preparation method (see Scheme 1):

CN101595092A (See Scheme 2).

Patent Document CN102762538A (see Scheme 3).
[0013]

CN104693114
Machine translated from chinese please bear with names
http://www.google.com/patents/CN104693114A?cl=en

Preparation Example 1 shell song in Spanish
 Under stirring, temperature 15 ~ 20 ° C,
was added dropwise 2mol / L tetrahydrofuran solution of
isopropylmagnesium chloride (available commercially available) 308ml (0 •
615mol, 5eq) to 2mol / L dimethylamine THF Solution (commercially
available can) 339ml (0.677mol, 5. 5eq) to give dimethylamine reaction
solution.
Under stirring, temperature 15 ~ 20 ° C,
was added dropwise 2mol / L tetrahydrofuran solution of
isopropylmagnesium chloride (available commercially available) 308ml (0 •
615mol, 5eq) to 2mol / L dimethylamine THF Solution (commercially
available can) 339ml (0.677mol, 5. 5eq) to give dimethylamine reaction
solution.
Under stirring, temperature 15 ~ 20 ° C, the compound of formula II 50. 0g (0 123mol, leq.) Was mixed with 500ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition continued at 25 The reaction was stirred for ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 700ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water; the filter cake was mixed by stirring with 500ml of acetone, the pH adjusted with triethylamine to 7-8; filtered; the cake at 40 ~ 45 ° C and dried under reduced pressure to give Pui Spanish song 45. 5g. . Yield: 82 0%; HPLC purity: 98.9%, of which 0.05% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Take the above Tony Qu Spanish 45.0g, at about 70 ° C under stirring dissolved in N, N- dimethylacetamide 180ml, a toluene solution of 360ml; cooling crystallization, filtration, the filter cake washed with an appropriate amount of acetone at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish HPLC purity 99.7%.
(+) LC-MS: m / z = 452 ([M + H] +). Che NMR (400MHz, DMS0-d6) S:…. 2 96 (s, 6H), 3 83 (s, 3H), 7. 06-7 09 (dd, 1H), 7. 55-7 59 ( m, 3H), 7. 80-7. 83 (dd, 1H), 8. 21-8. 23 (d, 1H), 8. 27-8. 30 (d, 2H), 8. 37-8. 40 (d, 1H), 8. 41-8. 43 (d, 1H), 10. 54 (br., 2H).
Preparation Example 2 Tony Qu Spanish maleate
Under stirring, temperature 0 ~ 5 ° C, dropping 2mol isopropyl magnesium chloride in tetrahydrofuran / L (available commercial available) 105ml (0 • 21mol, 8. 4eq) twenty methylamine hydrochloride 8. 91g (0 • llmol, 4. 4eq) in tetrahydrofuran 60ml of the suspension, the reaction solution obtained dimethylamine.
Under stirring, temperature 0 ~ 5 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, and then dropping the above reaction liquid dimethylamine; After the addition continued 10 The reaction was stirred for ~ 15 ° c, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 10 ~ 15 ° C, the reaction solution was added to an aqueous solution of 45g and 100ml dubbed maleic acid solution; the organic solvent was evaporated under reduced pressure and concentrated, filtered concentrate precipitated solid cake was washed with the right amount of water washing. Cake at 40 ~ 45 ° C and dried under reduced pressure to give Tony Qu Spanish maleate 12.lg. . Yield: 85 4%; HPLC purity: 98.6%, which is 0.03% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Take the above shellfish Spanish song maleate 10. 0g, at about 70 ° C under stirring dissolved in a mixed solvent of ethanol 50ml and 25ml of water, dropping water 150ml; cooling crystallization, filtration, the filter cake at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish maleate HPLC purity 99.9%.
: HNMR (400MHz, DMS〇-d6) 8: 3. 25 (s, 3H), 3. 32 (s, 3H), 3. 87 (s, 3H), 6. 02 (s, 2H) , 7. 19-7. 21 (dd, 1H), 7. 44-7. 45 (1H), 7. 75-7. 77 (d, 2H), 7. 97-9. 98 (d, 2H) , 8. 08-8. 13 (m, 3H), 8. 44-8. 45 (d, 1H), 9. 01 (br., 1H), 9. 37 (br., 1H), 11.04 (s , 1H), 11. 13 (s, 1H).
Preparation Example 3 Tony Spanish song of [0075] Example
Under stirring, temperature 25 ~ 30 ° C, isopropylmagnesium chloride in tetrahydrofuran was added dropwise a solution of 2mol / L (available commercially available) 81ml (0 • 161mol, 7eq) to 2mol / L dimethylamine THF Solution (commercially available can) 121ml (0 • 242mol, 10. 5eq) to give dimethylamine reaction solution.
Under stirring, temperature 25 ~ 30 ° C, the hydrochloride salt of a compound of formula II 10. 0g (0 023mol, leq.) Was mixed with 100ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition was complete The reaction continued stirring at 25 ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 210ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water; the filter cake with 90ml acetone was stirred and mixed, the pH adjusted with triethylamine to 7-8; filtration; cake was 45 ~ 50 ° C and dried under reduced pressure to give Pui Qu Spanish 8. 35g. Yield: 80.5%. HPLC purity: 98.7%, which is 0.03% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Preparation Example 4 shellfish Spanish song hydrochloride
Under stirring, temperature 15 ~ 20 ° C, dropping lmol / n-amyl magnesium bromide tetrahydrofuran solution (which can be commercialized available) 75ml (0 • 075mol, 3eq) to 2mol / L of dimethyl L amine in tetrahydrofuran (commercially available can) 56ml (0 • 113mol, 4. 5eq) to give dimethylamine reaction solution.
Under stirring, temperature 15 ~ 20 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition continued at 25 The reaction was stirred for ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 100ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water. Cake at 40 ~ 45 ° C and dried under reduced pressure to give Tony Qu Spanish hydrochloride 10.lg, yield:. 82 9%; HPLC purity: 99.0%, which is 0.02% dechlorination impurity VIII, from A impurities IX was not detected.
Take the above shellfish Spanish song hydrochloride 10. 0g, at about 70 ° C under stirring dissolved in N, N- dimethylacetamide 40ml, a toluene solution of 80ml; cooling crystallization, filtration, cake at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish hydrochloride HPLC purity 99.8%.
Preparation 5 shellfish Spanish song of [0082] Example
Under stirring, temperature 0 ~ 5 ° C, dropping lmol / diethyl zinc toluene solution of L (available commercially oriented) 50ml (0. 050mol, 2eq) to 2mol / L dimethylamine tetrahydrofuran (commercially available can) 28ml (0. 055mol, 2. 2eq) to give dimethylamine reaction solution.
Under stirring, temperature 0 ~ 5 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, and then dropping the above reaction liquid dimethylamine; After dropping 5 continues The reaction was stirred for ~ 10 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, in the next 5 ~ 10 ° C, the reaction mixture was added to about 2mol L dilute hydrochloric acid solution 70ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake was washed successively with a suitable amount of water; the filter cake with acetone l〇〇ml mixing, the pH adjusted with triethylamine to 7-8; filtered; the cake at 40 ~ 45 ° C under reduced pressed and dried to give Tony Qu Spanish 9. 03g. . Yield: 80 1%; HPLC purity: 99.0%, which is 0.02% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
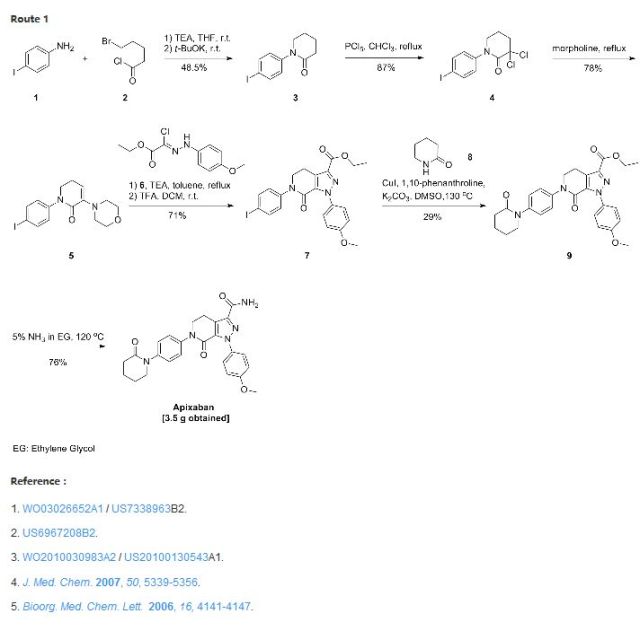
Preparation of compounds of Formula II Preparation Example 1
Methoxy-2-nitro – (5-chloro-pyridin-2-yl) -5 – benzamide (compound V) Preparation of [0086] (1) N-
 with stirring at room temperature,
5-methoxy-2-nitrobenzoic acid (Compound VI, can be commercially
available) 250g (1. 27mol, leq) and 2-amino-5-chloropyridine (Compound
VII .) 163g (l 27mol, leq) was suspended in 1700ml of acetonitrile,
pyridine 301g (3 81mol, 3eq), and then phosphorus oxychloride was added
dropwise 231g (l 52mol, 1 2eq);… After stirring for 1 hour the reaction
3500ml water quenching crystallization; the filter cake was washed with
water 1700mlX2; dried under reduced pressure to obtain compound V349g.
with stirring at room temperature,
5-methoxy-2-nitrobenzoic acid (Compound VI, can be commercially
available) 250g (1. 27mol, leq) and 2-amino-5-chloropyridine (Compound
VII .) 163g (l 27mol, leq) was suspended in 1700ml of acetonitrile,
pyridine 301g (3 81mol, 3eq), and then phosphorus oxychloride was added
dropwise 231g (l 52mol, 1 2eq);… After stirring for 1 hour the reaction
3500ml water quenching crystallization; the filter cake was washed with
water 1700mlX2; dried under reduced pressure to obtain compound V349g.
(2) 2-Amino -N- (5- chloro – pyridin-2-yl) -5-methoxy – benzamide (compound IV) is prepared
 with stirring at room temperature, the N-
(5- chloro – pyridin-2-yl) -5-methoxy-2-nitro – benzamide (Compound V)
300g (0 • 977mol, 1.Oeq) 3000ml was dissolved in acetic acid, and iron
powder was added portionwise 546g (9 77mol, 10eq.); After the addition
of iron stirring was continued for 3 hours, and then ethyl acetate and
water 6000ml 3000ml, liquid separation; the aqueous phase was separated
3000mlX2 extracted with ethyl acetate; combined organic phases were
washed with water, saturated aqueous sodium bicarbonate, saturated
sodium chloride solution, dried over anhydrous sodium sulfate, and
concentrated under reduced pressure to give compound IV244g.
with stirring at room temperature, the N-
(5- chloro – pyridin-2-yl) -5-methoxy-2-nitro – benzamide (Compound V)
300g (0 • 977mol, 1.Oeq) 3000ml was dissolved in acetic acid, and iron
powder was added portionwise 546g (9 77mol, 10eq.); After the addition
of iron stirring was continued for 3 hours, and then ethyl acetate and
water 6000ml 3000ml, liquid separation; the aqueous phase was separated
3000mlX2 extracted with ethyl acetate; combined organic phases were
washed with water, saturated aqueous sodium bicarbonate, saturated
sodium chloride solution, dried over anhydrous sodium sulfate, and
concentrated under reduced pressure to give compound IV244g.
(3) N- (5- chloro – pyridin-2-yl) -2- (4-cyano – benzoyl – amino) -5-methoxy – benzamide (compound II) is prepared
 at 10 ~ 20 ° C, a solution of a compound
of formula IV 200g (0 • 72mol, 1.Oeq) and triethylamine 109g (1. 08mol,
1. 5eq) 2000ml dissolved in tetrahydrofuran, to which was added dropwise
to cyano benzoyl chloride (compound III, commercially available
technology) 130g (0 79mol, 1.leq.) and tetrahydrofuran solution dubbed
1000ml, HPLC monitoring progress of the reaction; after the reaction was
filtered, the filter cake washed with an appropriate amount of ethanol,
dried under reduced pressure to obtain compound II263g. HPLC purity: 98.7%.
at 10 ~ 20 ° C, a solution of a compound
of formula IV 200g (0 • 72mol, 1.Oeq) and triethylamine 109g (1. 08mol,
1. 5eq) 2000ml dissolved in tetrahydrofuran, to which was added dropwise
to cyano benzoyl chloride (compound III, commercially available
technology) 130g (0 79mol, 1.leq.) and tetrahydrofuran solution dubbed
1000ml, HPLC monitoring progress of the reaction; after the reaction was
filtered, the filter cake washed with an appropriate amount of ethanol,
dried under reduced pressure to obtain compound II263g. HPLC purity: 98.7%.
(+) LC-MS: m / z = 407 ([M + H] +). Insect NMR (400MHz, DMS0-d6) S:… 3 85 (s, 3H), 7 16-7 .19 (dd, 1H), 7. 39-7 41 (d, 1H), 7. 93- 7. 96 (d, 2H), 8. 02-8. 04 (m, 4H), 8. 13-8. 14 (d, 2H), 8. 42-8. 43 (d, 1H), 11. 06 (br. 2H).
Example 2 Preparation of the hydrochloride salt of the compound of formula II
at 10 ~ 20 ° C, a solution of a compound of formula IV 40. 0g (0 • 14mol, 1.Oeq) was dissolved in 400ml of tetrahydrofuran, a solution of cyanobenzoyl chloride (Compound III, can be commercialized available) 24 8g (0 15mol, 1.leq) and tetrahydrofuran solution 200ml dubbed, HPLC monitoring progress of the reaction;.. After the reaction was filtered, the filter cake washed with ethanol and after an appropriate amount, and dried under reduced pressure to obtain a compound of formula II hydrochloride . HPLC purity: 99.5%.
Example 1: Preparation of Spanish Preparation and Form A half-L- malic acid shellfish song
At 55 ~ 60 ℃, the shellfish song Spanish 6.0g (13.3mmol), L- malic acid 1.1g (8.0mmol) was dissolved in tetrahydrofuran 70mL / water 7mL mixed solvent acetone was added with stirring 60mL, cooled to room temperature, Crystallization. Precipitated solid was filtered, and the resulting solid at 40 ~ 45 ℃ vacuum dried to give half L- malic acid shellfish Spanish song.
1H NMR(400MHz,MeOD)δ:2.355-2.419(dd,0.5H),2.735-2.781(dd,0.5H),3.226(s,6H),3.907(s,3H),4.302-4.326(dd,0.5H),7.195-7.224(dd,1H),7.448-7.455(d,1H),7.744-7.764(d,2H),7.821-7.849(dd,1H),8.145-8.165(d,2H),8.196-8.219(d,1H),8.238-8.261(d,1H),8.323-8.329(d,1H)。
Above 1 H-NMR results, δ: 3.907 (s, 3H) attributed to shellfish Spanish song molecule methyl CH 3 , 4.302-4.326 (dd, 0.5H) attributed to L- malic acid molecule methine CH , you can determine the song title product in shellfish Spanish and L- malic acid molar ratio of 2: 1.
Preparation of the compound of Formula II

8.0 parts) was prepared in a 780 L Hastelloy reactor (Reactor A) and adjusted to 0 0C (-3 to 3 0C). 2 M Dimethylamine in THF (161.0 kg, 5.0 eq.) and THF (63 kg, 2 parts) were charged into a 1900 L GLMS reactor (Reactor B) and adjusted to 0 0C (-3 to 3 0C) with maximum agitation. Hexyllithium (2.3 M, 97.2 kg, 4.5 eq.) was slowly charged to Reactor B while maintaining a max temperature of 10 0C. The pump and lines were rinsed forward to Reactor B with THF (3.2 kg). The Reactor B contents were adjusted to 0 0C (-3 to 3 0C), then transferred to Reactor A while keeping Reactor A temperature < 10 0C. The Reactor B pump and lines were rinsed forward with THF (31.4 kg, 1.0 part). The Reactor A contents were adjusted to 0 0C (-3 to 3 0C), and agitated at this temperature until the reaction was complete as verified by HPLC (1-2 hrs). After about 1 hr of agitation, in-process HPLC analysis indicated that 0 A% starting material remained (in-process criteria: max 1 A%). Reactor A contents were adjusted to -5 0C (-8 to -3 0C). In-process cleaning of Reactor B with water was performed. Two previously prepared aqueous solutions (NaHCO3 (35.0 kg, 1.1 parts) in water (236 kg, 7.5 parts), and Na2CO3 (35.0 kg 1.1 parts) in water (236 kg, 7.5 parts))were charged to Reactor B and adjusted to -3 0C (0 to 6 0C). Reactor A contents were transferred to Reactor B through an insulated line, maintaining the temperature of Reactor B at -8 0C to a maximum of 5 0C. The Reactor A pump and lines were rinsed forward with cold [-5 0C (-8 to -3 0C)] THF (31.4 kg, 1.0 part). Reactor B contents were adjusted to 22 0C (19-25 0C) and agitated for ca. 3 hrs. Slurry formation was visually confirmed, and Reactor B contents were filtered onto a 30″ centrifuge fitted with a filter cloth. The Reactor B pump and lines were rinsed forward onto the 30″ centrifuge fitted with a filter cloth with drinking water (63 kg, 2 parts). The wet filter cake (66.5 kg) was transferred back to Reactor B and submitted to a slurry wash in drinking water (1005 kg, 32 parts) at 22 0C (19-25) 0C for ca. 1 hr. The product was filtered onto the 30″ centrifuge (after in-process cleaning and fitting with a filter cloth), and the Reactor B lines and pump were rinsed forward with drinking water (63 kg, 2 parts). The water rinse was sampled for test by TDS, which was found to be 0.46%. The Reactor B pump, lines and wet filter cake were further rinsed with cold [0 0C (-3 to 3 0C)] ethanol (44 kg, 1.39 parts). The wet filter cake was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 35 0C. In-process LOD was 0% after ca. 24 hrs of drying, and the product was discharged (24.8 kg) in 76.7% yield. HPLC showed 98 % purity, with dechlorinated impurity at 1.14 %. Example 3
Preparation of the compound of Formula F Step 1. Synthesis of 2-nitro-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (C)

 To a 780 L Hastelloy reactor, compound C (33 kg, 1.0 eq.), 5%
platinum carbon (sulfided, 0.33 kg, 0.010 parts) and dichloromethane
(578 kg, 17.5 parts) were charged. Agitation was started and reactor
contents were adjusted to 22 0C (19-25 0C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 28 0C (25-31 0C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 28 0C (25 to 31 0C; maximum 31 0C)
until the reaction was complete by HPLC. After 16.5 hrs, the reaction
was deemed complete after confirming the disappearance of starting
material (0.472 A%). The contents of the reactor were circulated through
a conditioned celite pad (0.2-0.5 kg celite conditioned with 20-55 kg
dichloromethane) prepared in a 8″ sparkler filter to remove the platinum
catalyst. The reactor and celite bed were rinsed forward with two
portions of dichloromethane (83 kg, 2.5 parts each). The filtrate was
transferred to and concentrated in a 570 L GLMS reactor under a
atmospheric pressure to ca. 132 L (4 parts volume). Ethanol (69 kg, 2.1
parts) was charged and concentration continued under atmospheric
pressure to ca. 99 L (3 parts volume). In-process NMR indicated that the
dichloromethane content was 39%. Ethanol (69 kg, 2.1 parts) was charged
again and concentration continued again to ca. 99 L (3 parts volume).
In-process NMR indicated that the dichloromethane content was 5%. The
reaction mixture was then adjusted to 3 0C (0 to 6 0C),
agitated for ca. 1 hr, and the resulting slurry filtered onto a
jacketed pressure nutsche fitted with a filter cloth. The reactor, pump,
and lines were rinsed forward with cold [3 0C (0-6 0C)] ethanol (26 kg, 0.8 parts). The wet filter cake (36.6 kg) was dried under vacuum at 40-50 0C with a maximum temperature of water bath (to heat dryer jacket) of 50 0C.
LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The
dry product (D) was discharged (26.4 kg) in 89.5% yield. HPLC showed
98.4 A% purity, with dechlorinated impurity at 0.083 %. Step 3.
Synthesis of
N-(5-chloro-pyridin-2-yl)-2-(4-cyano-benzoyl-amino)-5-methoxy- benzamide
Hydrochloride (F)
To a 780 L Hastelloy reactor, compound C (33 kg, 1.0 eq.), 5%
platinum carbon (sulfided, 0.33 kg, 0.010 parts) and dichloromethane
(578 kg, 17.5 parts) were charged. Agitation was started and reactor
contents were adjusted to 22 0C (19-25 0C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 28 0C (25-31 0C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 28 0C (25 to 31 0C; maximum 31 0C)
until the reaction was complete by HPLC. After 16.5 hrs, the reaction
was deemed complete after confirming the disappearance of starting
material (0.472 A%). The contents of the reactor were circulated through
a conditioned celite pad (0.2-0.5 kg celite conditioned with 20-55 kg
dichloromethane) prepared in a 8″ sparkler filter to remove the platinum
catalyst. The reactor and celite bed were rinsed forward with two
portions of dichloromethane (83 kg, 2.5 parts each). The filtrate was
transferred to and concentrated in a 570 L GLMS reactor under a
atmospheric pressure to ca. 132 L (4 parts volume). Ethanol (69 kg, 2.1
parts) was charged and concentration continued under atmospheric
pressure to ca. 99 L (3 parts volume). In-process NMR indicated that the
dichloromethane content was 39%. Ethanol (69 kg, 2.1 parts) was charged
again and concentration continued again to ca. 99 L (3 parts volume).
In-process NMR indicated that the dichloromethane content was 5%. The
reaction mixture was then adjusted to 3 0C (0 to 6 0C),
agitated for ca. 1 hr, and the resulting slurry filtered onto a
jacketed pressure nutsche fitted with a filter cloth. The reactor, pump,
and lines were rinsed forward with cold [3 0C (0-6 0C)] ethanol (26 kg, 0.8 parts). The wet filter cake (36.6 kg) was dried under vacuum at 40-50 0C with a maximum temperature of water bath (to heat dryer jacket) of 50 0C.
LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The
dry product (D) was discharged (26.4 kg) in 89.5% yield. HPLC showed
98.4 A% purity, with dechlorinated impurity at 0.083 %. Step 3.
Synthesis of
N-(5-chloro-pyridin-2-yl)-2-(4-cyano-benzoyl-amino)-5-methoxy- benzamide
Hydrochloride (F)
 To a 780 L Hastelloy reactor, was charged 4-cyanobenzoyl chloride (E)
To a 780 L Hastelloy reactor, was charged 4-cyanobenzoyl chloride (E)
(17.2 kg, 1.1 eq.) and THF (92 kg, 3.5 parts). Reactor contents were agitated at 22 0C (19- 25 0C) until all of the solids had dissolved. The resulting solution was transferred to a lower receiver and the reactor was rinsed forward with THF (26 kg, 1 part). Compound D (26.4 kg, 1 eq.), THF (396 kg, 15 parts) and pyridine (2.90 kg, 0.4 eq.) were charged to a clean reactor. The pump and lines were rinsed forward with THF (34 kg, 1.3 parts). Via a metering pump, the 4-cyanobenzoyl chloride/THF solution was charged to the reactor, keeping the temperature at < 30 0C and rinsing forward with THF (ca. 10 kg). The resulting yellow-colored slurry was agitated at 22 0C (19-25 0C) for ca 2 hrs. In-process HPLC taken after 2 hrs showed a compound of Formula D content of 0%, indicating completion of the reaction. The slurry was filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, lines and wet cake were rinsed with three portions of ethanol (ca. 15 kg each). The wet filter cake was discharged (65.4 kg) and transferred back to the reactor for slurry wash in ethanol (317 kg, 12 parts) at 22 0C (19-25 0C) for ca. 1 hr. The slurry was filtered onto the pressure nutsche and the reactor, pump, lines, and wet filter cake were rinsed with two portions of ethanol (ca. 15 kg each) and two portions of THF (ca. 15 kg each). The wet filter cake was dried under vacuum with a maximum temperature of warm glycol bath (to heat the dryer jacket) of 40 0C. After 14.5 hrs of drying, LOD was 0.75%. The dried material was milled (screen 0.125″) to give 31.8 kg of product, which was dried under vacuum for another 10.5 hrs. LOD after drying was 1.8%, and the product was discharged (31.5 kg) in 74.8% yield (expected 60-90%). HPLC showed 100 % purity.
U.S. Patent No. 6,376,515 B2 discloses a class of benzamide based compounds as specific factor Xa inhibitors. In particular, U.S. Patent No. 6,376,515 B2 describes a compound identified as Example 206, which is also disclosed in U.S. Patent No. 6,835,739 B2 as Example 206 and herein identified as betrixaban, which has the chemical formula of Formula I:

Scheme 1

/////////////CN(C)C(=N)C1=CC=C(C=C1)C(=O)NC2=C(C=C(C=C2)OC)C(=O)NC3=NC=C(C=C3)Cl
4 EDOXABAN

Edoxaban, DU-176b
Edoxaban (DU-176b, trade names Savaysa, Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It was developed by Daiichi Sankyo and approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1] It was also approved by the FDA in January 2015 for the prevention of stroke and non–central-nervous-system systemic embolism.[2]
Daiichi Sankyo receives FDA approval for anti-clotting drug Savaysa
Japanese drug-maker Daiichi Sankyo has obtained approval from US Food and Drug Administration (FDA) for its anti-clotting drug Savaysa (edoxaban tablets)..8 JAN 2015

Daiichi Sankyo, APPROVED IN JAPAN as tosylate monohydrate salt in 2011 for the prevention of venous embolism in patients undergoing total hip replacement surgery



for synthesis see….http://www.sciencedirect.com/science/article/pii/S0968089613002642 Bioorganic & Medicinal Chemistry 21 (2013) 2795–2825, see s[pecific page 2808 for description ie 14/31 of pdf
WO 2010071121, http://www.google.com/patents/WO2010071121A1
WO 2007032498
N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide

Edoxaban (INN, codenamed DU-176b, trade name Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It is being developed by Daiichi Sankyo. It was approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1]
In animal studies, edoxaban is potent, selective for factor Xa and has good oral bioavailability.[2]
Daichi Sankyo’s edoxaban tosilate is an orally administered
coagulation factor Xa inhibitor that was approved and launched
in Japan for the preventive treatment of venous thromboembolic
events (VTE) in patients undergoing total knee arthroplasty, total
hip arthroplasty, or hip fracture surgery. Edoxaban has been
shown to have a rapid onset of anticoagulant effect due to short
Tmax (1–2 h) after dosing and sustained for up to 24 h post-dose.
Marketed under the brand name Lixiana, it is currently in phase
III studies in the US for the prevention of stroke and systemic embolic
events in patients with atrial fibrillation (AF) and venous
thromboembolism (VTE).
Several Phase II clinical trials have been conducted, for example for thromboprophylaxis after total hip replacement[3] (phase III early results compare well to enoxaparin[4]), and for stroke prevention in patients with atrial fibrillation[5][6].Those papers follow similar recent major trials showing similar results for the other new factor Xa inhibitors, rivaroxaban and apixaban.
A large phase III trial showed that edoxaban was non inferior to warfarin in preventing recurrent venous thromboembolic events with fewer episodes of major bleeding.[7]

http://www.google.com/patents/WO2014081047A1?cl=en
Chemically, edoxaban is
N1- (5-chloropyridin-2-yl) -N2- ( (IS, 2R/4S) -4- [ (dimethylamino) carbo nyl] -2- { [ ( 5-methyl-4 , 5,6, 7-tetrahydrothiazolo [5 , 4-c] pyridin-2-yl ) carbonyl] amino}eyelohexyl) ethanediamide , represented by the following formula (A) :
 (A) The p-toluenesulfonic acid monohydrate salt of compound A is represented b the following formula (B) :
(A) The p-toluenesulfonic acid monohydrate salt of compound A is represented b the following formula (B) :
 (B)
(B)
Edoxaban is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (also referred to as activated factor X or FXa) , and is useful as a preventive and/or therapeutic drug for thrombotic diseases.
Several processes are known in the literature for preparing edoxaban for example, U.S. Patent No. 7365205; U.S. Publication No . 20090105491.
U.S. Patent No. 7365205 provides a process for the preparation of edoxaban, wherein the process involves the use of
(IS, 4S, 5S) -4-iodo-6-oxabicyclo [3.2.1] octan-7-one, represented by the following formula (C) :
 (C)
(C)
as an intermediate.
The present inventors have identified that
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one, represented by the following formula (I) :
 ( I )
( I )
could also be used as an intermediate for the preparation of FXa inhibitory compounds like edoxaban. The present inventors have found that replacement of
(IS, 4S, 5S) -4-iodo-6-oxabicyclo [3.2.1] octan-7-one (C) with
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one (I) has a better atom economy and also an impact on cost.
A method for the synthesis of the
(IS, 4S, 5S) -4 -bromo- 6 -oxabicyclo [3.2.1] octan-7-one (I) was reported in Tetrahedron Letters, 51, (2010) Pages 3433-3435 which involves the reaction of ( IS) -cyclohex-3 -ene- 1-carboxylic acid represented by the following formula (II) :
 ( Π )
( Π )
with N-bromosuccinimide in the presence of molecular sieves using dichloromethane as a solvent. However, this reaction is carried out in dark over a period of 7 hours and does not provide a pure product .
Tetrahedron, Vol. 28, Pages 3393 -3399 , 1972 provides a process for the preparation of 4 -bromo- 6 -oxabicyclo [3.2.1] octan-7-one which involves the addition of 20% excess of a 2M solution of bromine in chloroform to a stirred solution of cyclohex- 3 -ene- 1-carboxylic acid (0.04 mol) in chloroform (250 mL) in the absence of a base . Extraction with aqueous sodium bicarbonate followed by acidification gave, after extraction with ether and evaporation of the extract, a mixture of cis & trans 3 , 4-dibromocyclohexanecarboxylic acid (6.7 g) and evaporation of the chloroform layer afforded the bromolactone (0.59 g) . It further provides a process for the preparation of
4 -bromo-6 -oxabicyclo [3.2.1] octan-7-one which involves the treating of cyclohex-3-ene-l-carboxylic acid (0.08 mol) dissolved in chloroform (450 mL) with 20% excess bromine in the presence of an equimolar amount of triethylamine (8.1 g) . After extraction of the amine with 2N hydrochloric acid, and work-up, bromolactone (10.7 g) and a mixture of cis & trans 3 , 4 -dibromocyclohexanecarboxylic acid (6.6 g) were obtained.
Tetrahedron Vol. 48, No. 3, Pages 539-544, 1992 provides a process for the preparation of
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one (I) which involves the addition of 1M solution of bromine in chloroform (30 mL) at 0°C to a solution of ( IS) -cyclohex-3 -ene- 1-carboxylic acid (0.024 mol) of formula (II) in chloroform (600 mL) in the presence of an equimolar amount of triethylamine (3.33 mL) . After work-up, the crude bromolactone obtained was recrystallized from petroleum ether.
However, bromination using bromine does not provide a pure product in good yield.
Heterocycles, Vol. 23, No. 8, Pages 2035-2039, 1985 provides a process for the 4-bromo-6-oxabicyclo [3.2.1] octan-7-one which involves the addition of cyclohex-3-ene-l-carboxylic acid (1.0 mM) in 1 , 2 -dimethoxyethane (2 mL) to a stirred solution of 90% Lead (IV) acetate (1.1 or 2.2 mM) in 1 , 2 -dimethoxyethane (4 mL) followed by the addition of Zinc bromide (2.2 mM) in 1 , 2 -dimethoxyethane (4 mL) and continuing the stirring for 10-30 minutes at 0°C . The reaction mixture was poured into a solution of ice-cold water (30 mL) and 10% hydrochloric acid (10 mL) , and extracted with ether (50 mL X 3) . The combined ether extract was washed successively with saturated sodium hydrogen carbonate solution (20 mL) , 10% sodium thiosulphate solution (5 mL) , and brine (10 mL) , and dried over sodium sulphate. Evaporation of the solvent gave crude lactone which were separated and purified (42% yield) . However, this reaction does not provide a pure product in good yield.
Heterocycles, Vol. 31, No. 6, Pages 987-991, 1990 provides a method for bromolactonization using a
dimethylsulfoxide-trimethylsilyl bromide-amine system. The bromolactonization is carried out for 10 to 72 hours using different solvents and triethylamine or diisopropylethyl amine as base. However, this process does not provide a product in high yield. Further the process afforded the cis isomer exclusively. Journal of the Chemical Society, Perkin Transactions 1:
Organic and Bio-Organic Chemistry (1972-1999) (1994) , (7) , Pages 847-851 provides a method for bromolactonization using a
dimethylsulfoxide-trimethylsilyl bromide-amine system. The bromolactonization is carried out for 12 hours using
dimethylsulfoxide and chloroform solvent system and triethylamine or diisopropylethyl amine as base. However, this process resulted in a low yield of about 55%. Citation List
Patent Literature
PTLl: U.S. Patent No. 7365205
PTL2: U.S. Publication No. 20090105491.
Non Patent Reference
NPLl: Feng Chen et al . , Tetrahedron Letters, 51, (2010) Pages 3433-3435.
NPL2 : G. Belluci et al . , Tetrahedron, Vol. 28, No. 13, Pages 3393-3399, 1972.
NPL3 : Marco Chini et al ., Tetrahedron Vol .48, No. 3, Pages 539-544 , 1992.
NPL4 : Y. Fujimoto et al . , Heterocycles , Vol. 23, No. 8, Pages 2035-2039, 1985.
NPL5: C. Iwata et al . , Heterocycles, Vol. 31, No. 6, Pages 987-991, 1990. -
NPL6 : K. Miyashita et al . , Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999) (1994) , (7) , Pages 847-851.
Summary of Invention
Technical Problem
It is an object of the present invention to solve the problems associated with the prior art, and to provide an improved and efficient method for the preparation of
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one of formula (I).
Solution to Problem As a result of conducting diligent studies to attain the object, the present inventors have found that: surprisingly, the use of N-bromosuccinimide or bromohydantoin (representative is
1, 3-dibromo-5, 5-dimethylhydantoin) as brominating agent in the presence of a base selected from calcium oxide or calcium hydroxide, in specific mole ratios in a solvent selected from the group consisting of dichloromethane , toluene, tetrahydrofuran, ethyl acetate, hexanes, cyclopentyl methyl ether (CPME) or a mixture thereof can efficiently produce a pure
( IS , 4S , 5S) -4 -bromo- 6 -oxabicyclo [3.2.1] octan- 7 -one (I) in better yields. The process provides obvious benefits with respect to economics, convenience to operate at a commercial scale.
................................
SEE
http://www.google.co.ug/patents/US20090105491
...............................
PATENT
http://www.google.com/patents/EP2589590A1?cl=en
FREE BASE
PATENT
http://www.google.com/patents/EP2589590A1?cl=en

Drug formulation , lixiana, edoxaban tosylate monohydrate, CAS 912273-65-5, C24 H30 Cl N7 O4 S . C7 H8 O3 S . H2 O, 738.274

OTHER SALTS

Edoxaban hydrochloride
CAS Number: 480448-29-1
Molecular Formula: C24H30ClN7O4S · HCl
Molecular Weight: 584.52 g.mol-1
Edoxaban is reported to be a member of the so-called “Xaban-group” and as such to be a low molecular inhibitor of the enzyme factor Xa, participating in the blood coagulation system. Therefore, edoxaban is classified as an antithrombotic drug and its possible medical indications are reported to be treatment of thrombosis and thrombosis prophylaxis after orthopaedic operations, such as total hip replacement, as well as for stroke prevention in patients with atrial fibrillation, the prophylaxis of the acute coronary syndrome and the prophylaxis after thrombosis and pulmonary embolism.
The IUPAC name for edoxaban is N’-(5-chloropyridin-2-yl)-N-[(15,2^,4S)-4- (dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[l ,3]thiazolo[5,4-c]pyridine-2- carbonyl)amino]cyclohexyl]oxamide. The chemical structure of edoxaban is shown in the formula (1) below:
 formula
( 1 ) While Edoxaban is reported to be soluble in strongly acidic
aqueous solutions, its solubility is considered to be very low in
neutral or alkaline aqueous media. EP 2 140 867 A 1 claims
an edoxaban-containing pharmaceutical composition comprising a
water-swelling additive and/or a sugar alcohol. Further, it is alleged
that compositions comprising lactose or cornstarch do not have good
dissolution properties. The claimed pharmaceutical compositions in EP 2
140 867 Al are considered to show good dissolution properties in a
neutral aqueous medium as well. Tablets comprising said composition were
produced by wet granulation. However, it turned out that prior art
pharmaceutical formulations comprising edoxaban being suitable for oral
administration are still improvable with regards to dissolution rate and
bioavailability. Further, stability and content uniformity of the known
formulations could be improved. Further, due to the intolerance of many
people to sugar alcohol(s), such as sorbitol, the use of sugar
alcohol(s) should be avoided.
UPDATE
formula
( 1 ) While Edoxaban is reported to be soluble in strongly acidic
aqueous solutions, its solubility is considered to be very low in
neutral or alkaline aqueous media. EP 2 140 867 A 1 claims
an edoxaban-containing pharmaceutical composition comprising a
water-swelling additive and/or a sugar alcohol. Further, it is alleged
that compositions comprising lactose or cornstarch do not have good
dissolution properties. The claimed pharmaceutical compositions in EP 2
140 867 Al are considered to show good dissolution properties in a
neutral aqueous medium as well. Tablets comprising said composition were
produced by wet granulation. However, it turned out that prior art
pharmaceutical formulations comprising edoxaban being suitable for oral
administration are still improvable with regards to dissolution rate and
bioavailability. Further, stability and content uniformity of the known
formulations could be improved. Further, due to the intolerance of many
people to sugar alcohol(s), such as sorbitol, the use of sugar
alcohol(s) should be avoided.
UPDATE

2-amino-5-methyl-4,5,6,7-tetrahydro thiazolone [5,4-c] pyridine
WO2015125710
WO2015125710
 UPDATE
UPDATE

1H NMR PREDICTION

.............
13 C NMR


![N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate NMR spectra analysis, Chemical CAS NO. 1229194-11-9 NMR spectral analysis, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate H-NMR spectrum](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fpic11.molbase.net%2Fnmr%2Fnmr_image%2F2015-01-11%2F001%2F717%2F1717235_1h.png&container=blogger&gadget=a&rewriteMime=image%2F*)
Edoxaban,
a factor Xa inhibitor, is supplied as edoxaban tosylate monohydrate.
The chemical name is
N-(5-Chloropyridin-2-yl)-N'-[(1S,2R,4S)-4-(N,N-dimethylcarbamoyl)-2-(5-methyl-
4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyridine-2-carboxamido)cyclohexyl]
oxamide mono (4- methylbenzenesulfonate) monohydrate. Edoxaban tosylate
monohydrate has the empirical formula C24H30ClN7O4S•C7H8O3S•H2O representing a molecular weight of 738.27. The chemical structure of edoxaban tosylate monohydrate is:
It
is a white to pale yellowish-white crystalline powder. The solubility
of edoxaban tosylate (pKa 6.7) decreases with increasing pH. It is
slightly soluble in water, pH 3 to 5 buffer, very slightly soluble at pH
6 to 7; and practically insoluble at pH 8 to 9.
SAVAYSA is available for oral administration as a 60 mg, 30 mg, or 15 mg round shaped, film-coated tablet, debossed with product identification markings. Each 60 mg tablet contains 80.82 mg edoxaban tosylate monohydrate equivalent to 60 mg of edoxaban. Each 30 mg tablet contains 40.41 mg edoxaban tosylate monohydrate equivalent to 30 mg of edoxaban. Each 15 tablet contains 20.20 mg edoxaban tosylate monohydrate equivalent to 15 mg of edoxaban.
The inactive ingredients are: mannitol, pregelatinized starch, crospovidone, hydroxypropyl cellulose, magnesium stearate, talc, and carnauba wax. The color coatings contain hypromellose, titanium dioxide, talc, polyethylene glycol 8000, iron oxide yellow (60 mg tablets and 15 mg tablets), and iron oxide red (30 mg tablets and 15 mg tablets).
5
APIXABAN

![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_vweznNjnvouZhDUK6sE9uaJZeFq3YzS22vBpOkp053HIEZlKTHE20815tD_qdj5dX4Mra5uhXzdk9cTcOKhiXHoB4pn5CQCr-dW5Tgk9-FCqDNnITZQXzL4g=s0-d) APIXABAN
APIXABAN
Apixaban
CAS 503612-47-3
APPROVALS
EMA————MAY 18, 2011
FDA…………………DEC28, 2012
PMDA………….. DEC25, 2012
CFDA………………JAN 22, 2013

Apixaban, sold under the tradename Eliquis, is a anticoagulant for the treatment of venous thromboembolic events. It is taken by mouth. It is a direct factor Xa inhibitor.
Apixaban was approved in Europe in 2012.[1] It was approved in the U.S. in 2014 for treatment and secondary prophylaxis of deep vein thrombosis (DVT) and pulmonary embolism (PE).[2] It is being developed in a joint venture by Pfizer and Bristol-Myers Squibb.[3][4]
Apixaban and other newer anticoagulants (dabigatran and rivaroxaban) appear equally effective as warfarin in preventing non-hemorrhagic stroke in people with atrial fibrillation and are associated with lower risk of intracranial bleeding.[7]
Apixaban was approved for the prevention of stroke in people with atrial fibrillation on December 28, 2012.[12] On March 14, 2014, it was approved for the additional use of preventing deep vein thrombosis and pulmonary embolism in people that had recently undergone knee or hip replacement.[13] On August 21, 2014, the FDA approved apixaban for the treatment of recurring deep vein thrombosis and pulmonary embolism.[2]
During development it was known as BMS-562247-01.

Thursday, August 21, 2014 – Bristol-Myers Squibb Company (NYSE: BMY) and Pfizer Inc. (NYSE: PFE) today announced the U.S. Food and Drug Administration (FDA) has approved a Supplemental New Drug Application (sNDA) for Eliquis for the treatment of DVT and PE, and for the reduction in the risk of recurrent DVT and PE following initial therapy. Combined, DVT and PE are known as VTE. It is estimated that every year, approximately 900,000 Americans are affected by DVT and PE.
http://www.drugs.com/newdrugs/fda-approves-eliquis-apixaban-deep-vein-thrombosis-pulmonary-embolism-4073.html?utm_source=ddc&utm_medium=email&utm_campaign=Today%27s+news+summary+-+August+21%2C+2014 –
See more at: http://worlddrugtracker.blogspot.in/2014/08/fda-approves-eliquis-apixaban-for.html
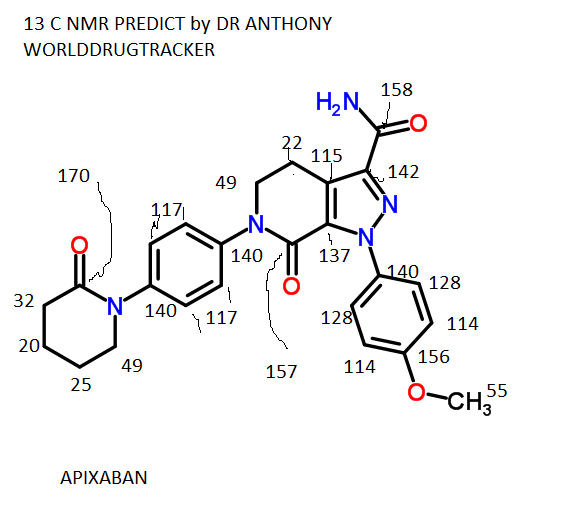
PREDICTIONS
1H NMR


13C NMR

COSY

1H NMR PREDICT


13 C NMR PREDICT



6
7
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
/////////////////////
2 RIVAROXABAN
RIVAROXABAN
5-Chloro-N-{[(5S)-2-oxo-3-[4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide
5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide
Molecular formula: C19H18ClN3O5S, MW435.9
CAS 366789-02-8
BAY 59-7939, XARELTO
Patent Expiration Date:
Feb 8, 2021(US7157456),
Dec 11, 2020(US7585860 and US7592339)
Originator and Manufacturer:Bayer
Marketer in the US: Johnson & Johnson
Sales: $1.3 billion (2013)
Originator and Manufacturer:Bayer
Marketer in the US: Johnson & Johnson
Sales: $1.3 billion (2013)
Rivaroxaban (BAY 59-7939) is an oral anticoagulant invented and manufactured by Bayer;[3][4] in a number of countries it is marketed as Xarelto.[1] In the United States, it is marketed by Janssen Pharmaceutica.[5] It is the first available orally active direct factor Xa inhibitor. Rivaroxaban is well absorbed from the gut and maximum inhibition of factor Xa
occurs four hours after a dose. The effects last approximately 8–12
hours, but factor Xa activity does not return to normal within 24 hours
so once-daily dosing is possible.
In September 2008, Health Canada granted marketing authorization for rivaroxaban for the prevention of venous thromboembolism(VTE) in people who have undergone elective total hip replacement or total knee replacement surgery.[8]
In September 2008, the European Commission granted marketing authorization of rivaroxaban for the prevention of venous thromboembolism in adults undergoing elective hip and knee replacement surgery.[9]
On July 1, 2011, the U.S. Food and Drug Administration (FDA) approved rivaroxaban for prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE), in adults undergoing hip and knee replacement surgery.[5]
On November 4, 2011, the U.S. FDA approved rivaroxaban for stroke prophylaxis in patients with non-valvular atrial fibrillation.
The drug compound having the adopted name "Rivaroxaban" has chemical name, 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-l,3-oxazolidin-5- yljmethyl)-2-thiophenecarboxamide; and has the structural formula I,
Formula I
The commercial pharmaceutical product XARELTO® tablets, contains rivaroxaban as active ingredient. Rivaroxaban is a factor Xa inhibitor useful as oral anticoagulant. Rivaroxaban can be used for the prevention and treatment of various thromboembolic diseases, in particular of deep vein thrombosis (DVT), pulmonary embolism (PE), myocardial infract, angina pectoris and restenoses after angioplasty or aortocoronary bypass, cerebral stroke,
transitory ischemic attacks, and peripheral arterial occlusive diseases.
U.S. Patent No. 7, 157,456 describes Rivaroxaban and process for the preparation thereof. The process of US '456 for rivaroxaban involves reaction of 2-[(2S)-2-oxiranylmethyl]-lH-isoindole-l,3(2H)-dione with 4-(4-aminophenyl)-3-morpholinone to provide 2-((2R)-2-hydroxy-3- { [4-(3-oxo-4-morpholiny)phenyl]amino Jpropyl)- lH-isoindole- 1 ,3(2H)-dione, which on cyclization using Ν,Ν-carbonyl diimidazole to afford 2-({5S)-2-Oxo-3-[4-(3-oxo-4-morpholiny)phenyl]-l,3-oxazolidin-5-yl}methyl)-lH-isoindole-l,3(2H)-dione, which on reacted with methylamine followed by reaction with 5-chlorothiophene-2-carbonyl chloride to provide Rivaroxaban.
Various processes for the preparation of rivaroxaban, its intermediates, and related compounds are disclosed in U.S. Patent Nos. 7,585,860; 7,351,823, 7,816,355, and 8,101,609; patent application Nos. WO 2011/012321, WO 2012/156983, WO 2012/153155, WO 2013/053739, WO 2013/098833, WO 2013/156936, WO 2013/152168, WO 2013/120464, WO 2013/164833, US 2012/0283434 and US 2013/184457; and J. Med. Chem. 2005, 48, 5900-5908.
In September 2008, the European Commission granted marketing authorization of rivaroxaban for the prevention of venous thromboembolism in adults undergoing elective hip and knee replacement surgery.[9]
On July 1, 2011, the U.S. Food and Drug Administration (FDA) approved rivaroxaban for prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE), in adults undergoing hip and knee replacement surgery.[5]
On November 4, 2011, the U.S. FDA approved rivaroxaban for stroke prophylaxis in patients with non-valvular atrial fibrillation.
The drug compound having the adopted name "Rivaroxaban" has chemical name, 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-l,3-oxazolidin-5- yljmethyl)-2-thiophenecarboxamide; and has the structural formula I,
Formula I
The commercial pharmaceutical product XARELTO® tablets, contains rivaroxaban as active ingredient. Rivaroxaban is a factor Xa inhibitor useful as oral anticoagulant. Rivaroxaban can be used for the prevention and treatment of various thromboembolic diseases, in particular of deep vein thrombosis (DVT), pulmonary embolism (PE), myocardial infract, angina pectoris and restenoses after angioplasty or aortocoronary bypass, cerebral stroke,
transitory ischemic attacks, and peripheral arterial occlusive diseases.
U.S. Patent No. 7, 157,456 describes Rivaroxaban and process for the preparation thereof. The process of US '456 for rivaroxaban involves reaction of 2-[(2S)-2-oxiranylmethyl]-lH-isoindole-l,3(2H)-dione with 4-(4-aminophenyl)-3-morpholinone to provide 2-((2R)-2-hydroxy-3- { [4-(3-oxo-4-morpholiny)phenyl]amino Jpropyl)- lH-isoindole- 1 ,3(2H)-dione, which on cyclization using Ν,Ν-carbonyl diimidazole to afford 2-({5S)-2-Oxo-3-[4-(3-oxo-4-morpholiny)phenyl]-l,3-oxazolidin-5-yl}methyl)-lH-isoindole-l,3(2H)-dione, which on reacted with methylamine followed by reaction with 5-chlorothiophene-2-carbonyl chloride to provide Rivaroxaban.
Various processes for the preparation of rivaroxaban, its intermediates, and related compounds are disclosed in U.S. Patent Nos. 7,585,860; 7,351,823, 7,816,355, and 8,101,609; patent application Nos. WO 2011/012321, WO 2012/156983, WO 2012/153155, WO 2013/053739, WO 2013/098833, WO 2013/156936, WO 2013/152168, WO 2013/120464, WO 2013/164833, US 2012/0283434 and US 2013/184457; and J. Med. Chem. 2005, 48, 5900-5908.
PAPER CONTAING SPECTRAL DATA
JOURNAL OF CHEMICAL RESEARCH v 35, issue 7, pg 400-4-1, 2011
An approach to the anticoagulant agent rivaroxaban via an isocyanate-oxirane cycloaddition promoted by MgI2.etherate
Chao Lia, Yingshuai Liua, Yongjun Zhangb and Xingxian Zhanga*
a College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310032, P. R. China
b Zhejiang Apeloa Medical Technology Co., Ltd, Dongyang 322118, P. R. China
a College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310032, P. R. China
b Zhejiang Apeloa Medical Technology Co., Ltd, Dongyang 322118, P. R. China
A convergent and efficient synthesis of anticoagulant rivaroxaban was developed using the cycloaddition of commercially
available (R)-epichlorohydrin with 4-(morpholin-3-one)phenyl isocyanate catalysed by MgI2 etherate as the
key step, in 22% overall yield.
Keywords: (R)-epichlorohydrin, isocyanate, MgI2.etherate, rivaroxaban
available (R)-epichlorohydrin with 4-(morpholin-3-one)phenyl isocyanate catalysed by MgI2 etherate as the
key step, in 22% overall yield.
Keywords: (R)-epichlorohydrin, isocyanate, MgI2.etherate, rivaroxaban
* Correspondent. E-mail: mhmosslemin@yahoo.com
(Rivaroxaban) (1):1
rivaroxaban 1 (689 mg) in 88% yield, Rf = 0.30 (ethyl acetate), as a white solid,
m.p. 229.3–230.7 °C(lit.1, 230 °C).
[α]D20 = −37° (c = 0.5, DMSO) [lit.1, [α]D21 = –38°(c = 0.2985, DMSO)].
IR (KBr) (νmax /cm−1): 3343, 1724 (C=O), 1649(C=O), 1523, 1430, 808, 756
δH
3.60–3.62 (m, 2H), 3.71–3.73 (m,2H), 3.84–3.87 (dd, J = 6.5, 9.5 Hz,
1H), 3.96–3.98 (m, 2H), 4.20 (s,2H), 4.18–4.21 (m, 1H), 4.83–4.86 (m,
1H), 7.20 (d, J = 4.0 Hz, 1H),7.41 (d, J = 9.0 Hz, 2H), 7.56 (d, J = 9.0
Hz, 2H), 7.69 (d, J = 4.0 Hz,1H), 8.99 (t, J = 5.5 Hz, 1H).
δC 42.19, 47.43, 49.00, 63.46, 67.71,71.30, 118.35, 125.92, 128.11, 128.43, 133.24, 136.48, 137.08,138.43, 154.08, 160.79, 165.95.
LIT
REF 1=S. Roehrig, A. Straub, J. Pohlmann, T. Lampe, J. Pernerstorfer,
K.Schlemmer, P. Reinemer and E. Perzborn, J. Med. Chem., 2005, 48, 5900.
STRUCTURE


SIMILARITY
Rivaroxaban bears a striking structural similarity to the antibiotic linezolid: both drugs share the same oxazolidinone-derived core structure. Accordingly, rivaroxaban was studied for any possible antimicrobial effects and for the possibility of mitochondrial toxicity,
which is a known complication of long-term linezolid use. Studies found
that neither rivaroxaban nor its metabolites have any antibiotic effect
against Gram-positive bacteria. As for mitochondrial toxicity, in vitro studies found the risk to be low
IH NMR PREDICT
13 C NMR PREDICT
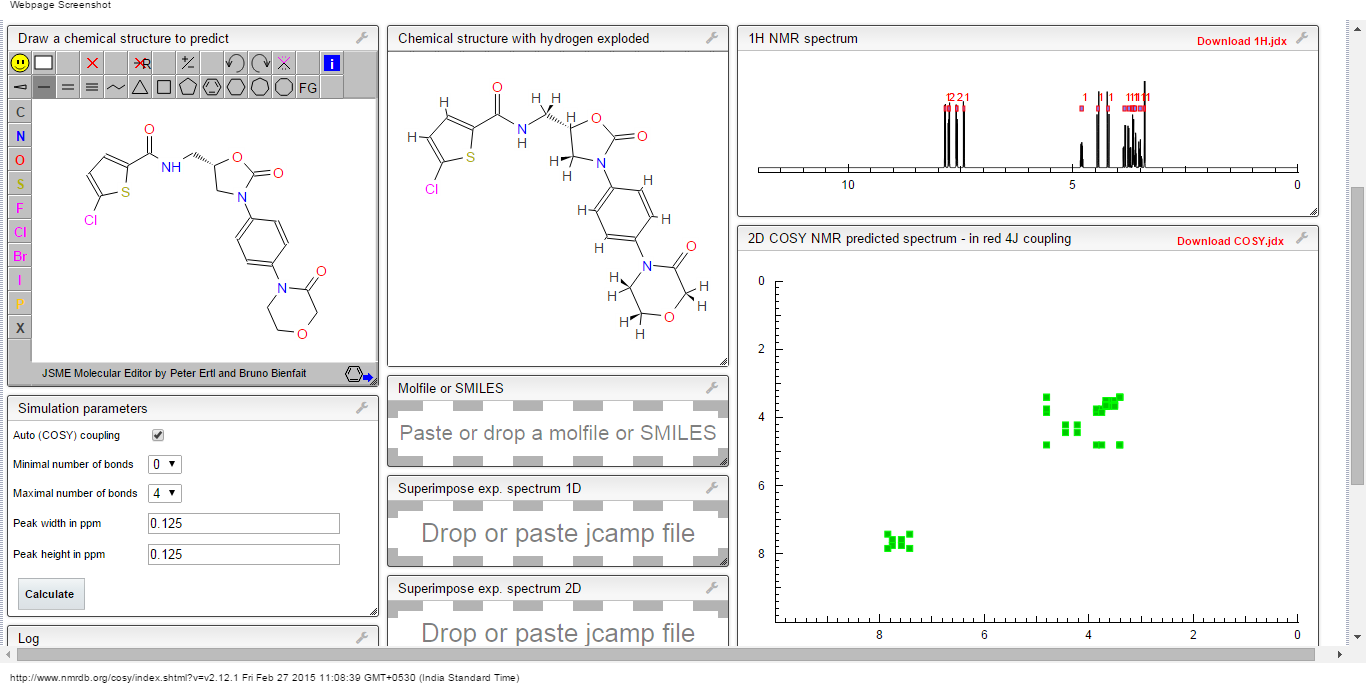
COSY NMR.
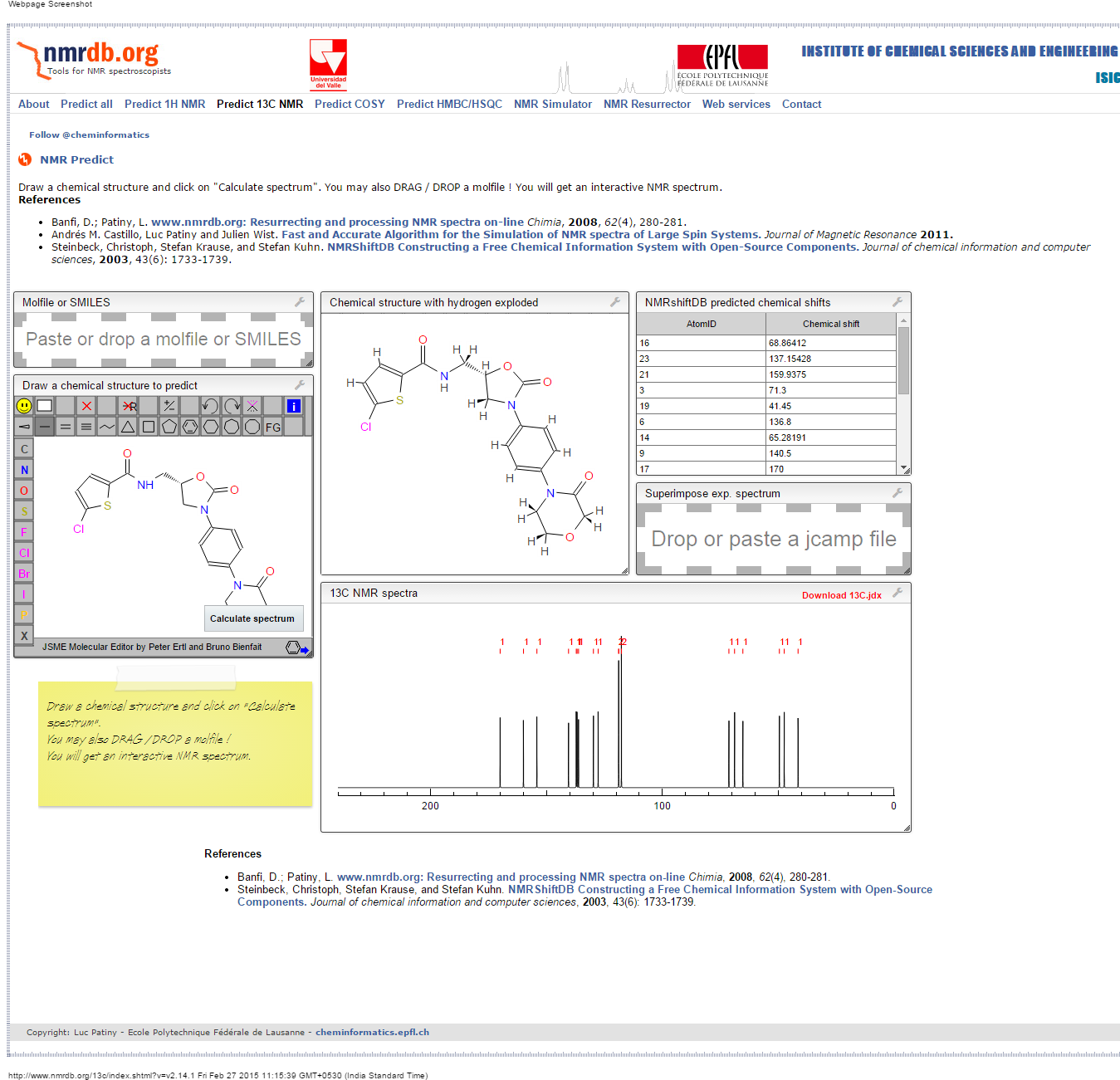
13 C NMR.....http://www.nmrdb.org/13c/index.shtml?v=v2.14.1
CLICK TO PREDICT..ALLOW SOME TIME TO LOAD ON NMRDB SITE.....CHECK JAVA AND FLASH SETTINGS
ABOVE PICTURES ARE THE ONES YOU WILL GET
New patent WO-2015104605
Process for preparing rivaroxaban - comprising the reaction of a thioester compound and its salts with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one.Wockhardt Ltd
The synthesis of (II) via intermediate (I) is described (example 7, page 15)
4-{4-[(5S)-5-(Aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one (formula III) is (I) and rivaroxaban is (II) (claim 1, page 16).
The present invention relates to a process for the preparation of Rivaroxaban and its novel intermediates, or pharmaceutically acceptable salts thereof. The present invention provides novel intermediates, which may be useful for the preparation of Rivaroxaban or its pharmaceutically acceptable salts thereof. The process of preparation by using novel intermediate is very simple cost effective and may be employed at commercial scale. The product obtained by using novel intermediate yield the Rivaroxaban of purity 99% or more, when measured by HPLC. The present invention especially relates to a process for the preparation of Rivaroxaban from thioester of formula II, or a pharmaceutically acceptable salt thereof, wherein R is leaving group.
process includes the step of , reacting thioester of formula IIA or pharmaceutically acceptable salt thereof
Formula IIA
with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one of formula III,
Formula III
Formula I
EXAMPLE 7: One pot process for Rivaroxaban
The triphenylphosphine (11.5g) and mercaptobenzothiazole disulphide (15.31g) were taken in methylene chloride and reaction mixture was stirred at 28°C -30°C for 1 hr. The 5-chlorothiophene-2-carboxylic acid (7.2g) and triethylamine (3.8 g) were added to the above reaction mixture. The reaction mixture is stirred at 0°C -25 °C for 1 hr. after 1 hr 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one (lOg) and triethylamine (3.8g) were added. The resulting reaction mixture further stirred for 2 hrs. After completion of the reaction, water was added and stirred for 10 min. aqueous layer was separated and washed with methylene chloride. The organic layer was acidified to pH 6-7 with 2N hydrochloric acid and finally the organic layer was concentrated to get desired product. The product was purified and dried to yield Rivaroxaban.
Yield: 10.0 gm
Purity: 99.3 %
EXAMPLE 8: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent ethyl acetate and base potassium hydroxide were used to get the rivaroxaban.
EXAMPLE 9: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent acetonitile and base potassium carbonate were used, methylene chloride was added in the reaction mixture to extract the Rivaroxaban.
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015104605&recNum=7&maxRec=57790&office=&prevFilter=%26fq%3DOF%3AWO%26fq%3DICF_M%3A%22C07D%22&sortOption=Pub+Date+Desc&queryString=&tab=PCTDescription
..............
http://www.google.com/patents/WO2013120465A1?cl=en
Rivaroxaban,
chemically
(S)-5-chloro-N-({2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-l,3-
oxazolidin-5-yl}methyl)thiophene-2-carboxamide, described by formula
(1), was developed by the company Bayer Healthcare (WO 01/47919, 2001).
Rivaroxaban is applied in the clinical practice as the active ingredient
of an orally available anticoagulant that is commercially marketed as
Xarelto and is used in the prevention and treatment of arterial or
venous thromboembolic disorders. In its effect, rivaroxaban is
characterized by direct selective inhibition of the FXa coagulation
enzyme (Drugs of the Future 2006, 31(6): 484-493).
H2

For
the preparation of rivaroxaban several key structures, referred to as
building blocks, can be used as advanced intermediates. Virtually all
the so far described syntheses are using two such building blocks. The
first one are derivatives of 4-(4-aminophenyl)morpholin-3-one, where it
may be the case of an unsubstituted amine (2, G means hydrogen), or a
derivative alkylated on nitrogen, or a carbamate derived from this
compound (2, G means an alkyl or COOalkyl group). The other general and
commonly used building block for the rivaroxaban molecule are
derivatives of 5-chlorothiophene-2-carboxylic acid (3, X means -OH), or
its functional derivatives such as the chloride and amide (3, X means
-CI or -NH2). Various synthetic approaches used for synthesis
of rivaroxaban differ from each other mainly as regards the chiral
building block, which is the source for the construction of the central
heterocycle, i.e., 2-oxo-l,3-oxazolidine, wherein the chirality centre
is also located. For pharmaceutical purposes one optical isomer derived
from rivaroxaban is only used, in particular the target molecule with
the absolute configuration (5)-. The selection of a suitable chiral
building block must be subjected to this fact.

(4) (5) (6) (7) (8)
Chiral
building blocks that have been successfully used for synthesis of
rivaroxaban include (5)-glycidyl phthalimide (4),
(S)-3-aminopropane-l,2-diol (5), ( ?)-epichlorohydrin (6) and
(i?)-glycidyl butyrate (7). (S)-glycidol (8) was used as a starting
material for the preparation of (5)-glycidyl phthalimide (4)
(Tetrahedron: Asymmetry, Vol. 7, No. 6, pp. 1641-1648, 1996).
The
known methods of chemical synthesis of rivaroxaban (1) are described in
Schemes 1 to 7. The first one is the process according to Scheme 1 (WO
01/47919 Bayer, US 7 157 456 B2, J.MedChem. (2005), 48(19), 5900-5908),
which starts from 4-(4-aminophenyl)morpholin-3- one and (5)-glycidyl
phthalimide (4). The second synthetic process follows Scheme 2 (WO
2004/060887, Bayer) and starts from 5-chlorothiophene-2-carboxylic acid
(3, X means -OH) and (5)-3-aminopropane-l,2-diol (5).
4-(4-aminophenyl)morpholin-3-one only engages in the synthesis in the
penultimate stage in case of the process according to Scheme 2.

Scheme 1

The
third synthetic process, which proceeds according to Scheme 3, was
mainly used for preparation of deuterated analogs of rivaroxaban (WO
2009/023233 Al, Concert Pharm.). It also represents the first synthetic
process in which (i?)-epichlorohydrin (6) was used as the chiral
building block. The other key starting material for the third process
was 4-(4- aminophenyl)morpholin-3-one. The fourth synthetic process,
which proceeds according to Scheme 4 (WO 2010/124835 Al, Apotex), again
uses (i?)-epichlorohydrin as the chiral building block, which reacts
with the alkyl carbamate derived from 4-(4- aminophenyl)morpholin-3-one
in the key stage. The fifth synthetic process, which proceeds according
to Scheme 5 (US 20110034465 Al), also uses (i?)-epichlorohydrin as the
chiral building block, which directly reacts with
4-(4-aminophenyl)morpholin-3-one in the key stage, which is the same
reaction as in the third process. The differences between the third and
fifth processes consist in the preparation method of the
2-oxo-l,3-oxazolidine cycle and in the carbonylation agent used. While
the third process uses Ι,Γ-carbonyldiimidazol (CDI) as the carbonylation
agent, the fifth process uses more available and cheaper alkyl
chloroformates.

Scheme 3

Scheme 4

Scheme 5
The
sixths synthetic process, which proceeds according to Scheme 6 (WO
2011/080341 Al), uses (7?)-glycidyl butyrate (7) as the chiral building
block, which in the key stage reacts with the alkyl carbamate derived
from 4-(4-aminophenyl)morpholin-3-one. The last, seventh synthetic
process leading to rivaroxaban proceeds according to Scheme 7 (WO 201
1/098501 Al) and, like process 2, uses (S)-3-aminopropane-l,2-diol (5)
as the chiral building block. The differences between the second and
seventh processes consist in the preparation process of the
2-oxo-l,3-oxazolidine cycle and the carbonylation agent used. While the
second process uses Ι,Γ-carbonyldiimidazol (CDI) as the carbonylation
agent, the fifth process uses the cheaper, but very toxic phosgene.

Scheme 6

Scheme
7 The processes used for the synthesis of rivaroxaban differ from each
other especially in the chiral building block (compounds 4 to 7) and in
the carbonylation agents (CDI, alkyl chloroformates, phosgene) used.
Another difference can be found in the method of performing deprotection
reactions, i.e. such reactions that lead to elimination of the
protecting groups, initially bound to the nitrogen atom of the advanced
intermediates and which had the initial purpose of protecting these
intermediates from undesired chemical transformations. No deprotection
reactions were necessary in the case of the processes according to
Schemes 2, 4 and 7, as the protecting groups bound to the nitrogen atom
eventually became part of the final product. In the case of process 6 it
was necessary to deprotect the fert-butyl group bound to the nitrogen.
The reaction used was an acid catalyzed reaction of the tert-butyl
group, releasing isobutylene according to Scheme 8. In normal conditions
isobutylene is a gas and thus can be very easily separated from the
final product.

isobutylen

.................
WO 01/47919 discloses ー species from 4_ (4_ aminophenyl) -3_ morpholinone (I) Preparation of rivaroxaban approach:
..............
US 07/149522 discloses ー kind to 5_ chlorothiophenes _2_ carbonyl chloride (IV) is a method for preparing raw rivaroxaban in:
.............
http://www.google.com/patents/CN102786516A?cl=en

The 11.6 g (37.0 mmol) 4- {4 - [(5S) -5- (aminomethyl) -2-oxo-1,3-oxazolidin-3-yl] phenyl} morpholin-3 -one hydrochloride was added 40ml of water, was added 4. 64 g (43 8 mmol.) Na2CO3 stirred and dissolved; then added 50 ml of toluene, was added dropwise at 10 ° C under the mixture, the mixture is 8. 0 g ( 44. 4 mmol) 5- chloro-thiophene-2-carbonyl chloride was dissolved in 15 ml of toluene, 20 min the addition was complete, then stirring was continued at room temperature, TLC monitoring progress of the reaction, 2 h after completion of the reaction; and the filter cake washed with water and washed with acetone to give a pale yellow solid 19. 6 g, used directly ko acid recrystallization, as a white solid 15. 2 g,
mp 227. 2 - 228. 1 ° C, [a] D21 = -38 2 ° (. c = 0. 30, DMS0), rivaroxaban yield of 94%, the total yield of 87.5% 0
1H-NMR (DMSO) 8: 3. 61 (. 2 H, t, / = 5 4 Hz), 3. 71 (2 H, t, / = 5 4 Hz.), 3.85 (IH, m ), 3.97 (2 H, t, J = 4. 5 Hz), 4. 19 (3 H, t, / = 7. 5 Hz), 4.84 (IH, m), 7. 19 (IH, d, / = 4. 2Hz), 7.40 (2 H, d, /=9.0 Hz), 7. 57 (2 H, t, /=9.0 Hz), 7. 69 (IH, d, J = 4. 19 Hz), 8. 96 (IH, t, / = 5. 7 Hz).
.....................
WO2013120465
EXAMPLE 28 (preparation of rivaroxaban)
1H NMR (250 MHz, DMSO-D6), δ (ppm): 3.61 (t, 2H, CH2); 3.71 (m, 2H, CH2); 3.85 and 4.19 (m, 2x1 H, CH2); 3.97 (m, 2H, CH2); 4.19 (s, 2H, CH2); 4.84 (pent, 1H, CH); 7.18 (d, 1H); 7.40 (m, 2H); 7.56 (m, 2H); 7.68 (d, 1H); 8.95 (bt, 1H, NH).
13C NMR (250 MHz, DMSO-D6), δ (ppm): 42.2; 47.4; 49.0; 63.4; 67.7; 71.3; 1 18.3; 125.9; 128.1 ; 128.4; 133.2; 136.4; 137.0; 138.4; 154.0; 160.8; 165.9.
MS (m/z): 436.0729 (M+H)+. ation)

..........................

........................
5- chloro-thiophene-2-chloride by condensation, bromide, with 4- (4-amino-phenyl) -3-morpholinone cyclization reaction rivaroxaban, the following reaction scheme :( References : W02005068456, US20070149522, DE10300111)

...........................
5-
chloro-thiophene-2-chloride by condensation, oxidation, and 4-
(4-amino-phenyl) -3-morpholinone cyclization reaction racemic
rivaroxaban, since the epoxidation step is not give any
stereoselectivity, the final chiral separation need to get rivaroxaban,
the reaction scheme is as follows :( References: W0-0147919)


............
4- (4- amino-phenyl) -3-morpholinone by condensation, cyclization, and potassium phthalimide after reaction with methyl chloroformate to give (S) -2 - hydroxy -3- (I, 3- dioxo - isoindoline-2-yl) propyl-4- (3-oxo --morpholino) phenyl carbamate, by condensation, methylamine and Ethanol action under profit rivaroxaban, the following reaction scheme (Ref: US20110034465):
..........
4- (4- amino-phenyl) -3-morpholinone (R) and - epichlorohydrin, in the DMF solvent phthalimide potassium salt was reacted with ammonia solution and then prepared to succeed amino compound, and 5-chloro-thiophene-2-chloride in pyridine catalyzed system benefit rivaroxaban, the following reaction scheme (Ref: W02009023233):
.............
4- (4- amino-phenyl) -3-morpholinone after condensation with (R) - epichlorohydrin, then the 5-chloro-thiophene-2-amide lithium chloride and tert-butyl the reaction of an alcohol potassium enrichment rivaroxaban, the following reaction scheme (Ref: US7816355):
...................
3-chloro-1,2-propanediol by cyclization, the reaction with phthalimide, then with 4- (4-aminophenyl) -3-morpholinone reaction, CDI and hydrazine to give 4- {4- [(5S) -5- (aminomethyl) -2-oxo-1,3-oxazolidin-3-yl] phenyl} morpholin-3-one under the influence, in pyridine and under the action of tetrahydrofuran and 5-chloro-thiophene-2-chloride benefit rivaroxaban, the following reaction scheme (Reference: Gutcait, A. et al Tetrahedron:.. Asymmetry 1996, 7 (6), 1641-1648 Roehrig, .. S. et al J. Med Chem 2005,48 (19), 5900-5908)..:
..............
http://www.google.com/patents/CN102702186A?cl=zh

[0072] Method One:

I. 85mmol) added to the reaction flask, stirred at room temperature for 10 minutes, cooled to 0 ° C, a solution of 5-chloro-2-thiophene chloride (224mg, 1.24mm0l), stirred at room temperature overnight; after the completion of the reaction, spin dry, rinse with anhydrous alcohol ko, filtered, washed ko anhydrous alcohol three times to obtain a white solid product rivaroxaban (215mg, embodiments of the total yield of 7,8 80%).
[0075] 1H-Mffi (DMSC) JOOMHz, δ d m):.... 3 61 (t, 2H, J = 5 6Hz), 3. 71 (t, 2H, J = 5 2Hz), 3 89 ( m, 1H), 3. 97 (t, 2H, J = 4. 4Hz), 4. 20 (m, 3H), 4. 85 (m, 1H), 7. 18 (d, 1H, J = 4. 0Hz), 7. 40 (d, 2H, J = 8. 8Hz), 7. 56 (d, 2H, J = 8. 8Hz), 7. 73 (d, 1H, J = 4. 0Hz).
The method of writing is:

SYNTHESIS
SYN 1
BAYER HEALTHCARE AG Patent: WO2004/60887 A1, 2004 ; Location in patent: Page/Page column 8; 10-11 ;
SYN 2
MEDICHEM S.A.; MANGION, Bernardino; DURAN LOPEZ, Ernesto Patent: WO2012/35057 A2, 2012 ; Location in patent: Page/Page column 34 ;
SYN 3
EGIS GYOGYSZERGYAR NYILVANOSAN MUeKOeDOe RESZVENY-TARSASAG; SIPOS, Eva; KOVANYINE LAX, Gyoergyi; HAVASI, Balazs; VOLK, Balazs; KRASZNAI, Gyoergy; RUZSICS, Gyoergy; BARKOCZY, Jozsef; TOTHNE LAURITZ, Maria; LUKACS, Gyula; BOZA, Andras; HEGEDUeS, Laszlo Jozsef; TABORINE TOTH, Maria Julia; PECSI, Eva Patent: WO2012/153155 A1, 2012 ; Location in patent: Page/Page column 49 ;
SYN4
EGIS GYOGYSZERGYAR NYILVANOSAN MUeKOeDOe RESZVENY-TARSASAG; SIPOS, Eva; KOVANYINE LAX, Gyoergyi; HAVASI, Balazs; VOLK, Balazs; KRASZNAI, Gyoergy; RUZSICS, Gyoergy; BARKOCZY, Jozsef; TOTHNE LAURITZ, Maria; LUKACS, Gyula; BOZA, Andras; HEGEDUeS, Laszlo Jozsef; TABORINE TOTH, Maria Julia; PECSI, Eva Patent: WO2012/153155 A1, 2012 ; Location in patent: Page/Page column 68 ;
SYN 5
INTERQUIM, S.A.; Berzosa Rodríguez, Xavier; Marquillas Olondriz, Francisco; Llebaria Soldevilla, Amadeo; Serra Comas, Carme Patent: US2014/128601 A1, 2014 ; Location in patent: Paragraph 0068 ;
SYN 6
MEGAFINE PHARMA (P) LTD; MATHAD Vijayavitthal Thippannachar; PATIL NILESH SUDHIR, Nilesh; NIPHADE NAVNATH CHINTAMAN, Navnath; MALI ANIL CHATURLAL, Anil; BODAKE MAHENDRA BHAGIRATH, Mahendra; IPPAR SHARAD SUBHASH, Sharad; TALLA RAJESH, Rajesh Patent: WO2013/121436 A2, 2013 ; Location in patent: Page/Page column 31 ;
References
- "Xarelto: Summary of Product Characteristics". Bayer Schering Pharma AG. 2008. Retrieved 2009-02-11.
- Abdulsattar, Y; Bhambri, R; Nogid, A (May 2009). "Rivaroxaban (xarelto) for the prevention of thromboembolic disease: an inside look at the oral direct factor xa inhibitor.".P & T : a peer-reviewed journal for formulary management 34 (5): 238–44.PMID 19561868.
- Roehrig S, Straub A, Pohlmann J et al. (September 2005). "Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): an oral, direct factor Xa inhibitor". Journal of Medicinal Chemistry 48 (19): 5900–8. doi:10.1021/jm050101d.PMID 16161994.
- Perzborn, Elisabeth; Roehrig, Susanne; Straub, Alexander; Kubitza, Dagmar; Misselwitz, Frank (17 December 2010). "The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor". Nature Reviews Drug Discovery 10 (1): 61–75. doi:10.1038/nrd3185.
- "FDA Approves XARELTO® (rivaroxaban tablets) to Help Prevent Deep Vein Thrombosis in Patients Undergoing Knee or Hip Replacement Surgery" (Press release).Janssen Pharmaceutica. 2011-07-01. Retrieved 2011-07-01.
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). "Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.". Thrombosis 2013: 640723. doi:10.1155/2013/640723. PMC 3885278.PMID 24455237.
- Brown DG, Wilkerson EC, Love WE (March 2015). "A review of traditional and novel oral anticoagulant and antiplatelet therapy for dermatologists and dermatologic surgeons".Journal of the American Academy of Dermatology 72 (3): 524–34.doi:10.1016/j.jaad.2014.10.027. PMID 25486915.
- "Bayer's Xarelto Approved in Canada" (Press release). Bayer. 2008-09-16. Retrieved2010-01-31.
- "Bayer’s Novel Anticoagulant Xarelto now also Approved in the EU" (Press release).Bayer. 2008-02-10. Retrieved 2010-01-31.
- "Medication Guide--Xarelto" (PDF). http://www.fda.gov/. U.S. Food and Drug Administration. Retrieved 1 September 2014.
- "Xarelto Side Effects". http://www.webmd.com/. WebMD. Retrieved 1 September2014.
- "Xarelto Side Effects Center". http://www.rxlist.com/. RxList. Retrieved 1 September2014.
- Eriksson BI, Borris LC, Dahl OE et al. (November 2006). "A once-daily, oral, direct Factor Xa inhibitor, rivaroxaban (BAY 59-7939), for thromboprophylaxis after total hip replacement". Circulation 114 (22): 2374–81.doi:10.1161/CIRCULATIONAHA.106.642074. PMID 17116766.
- Eriksson BI, Borris LC, Friedman RJ et al. (June 2008). "Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty". The New England Journal of Medicine 358(26): 2765–75. doi:10.1056/NEJMoa0800374. PMID 18579811.
- Kakkar AK, Brenner B, Dahl OE et al. (July 2008). "Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial". Lancet 372 (9632): 31–9.doi:10.1016/S0140-6736(08)60880-6. PMID 18582928.
- Lassen MR, Ageno W, Borris LC et al. (June 2008). "Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty". The New England Journal of Medicine358 (26): 2776–86. doi:10.1056/NEJMoa076016. PMID 18579812.
- Turpie A, Bauer K, Davidson B et al. "Comparison of rivaroxaban – an oral, direct factor Xa inhibitor – and subcutaneous enoxaparin for thromboprophylaxis after total knee replacement (RECORD4: a phase 3 study) / European Federation of National Associations of Orthopaedics and Traumatology Annual Meeting; May 29 – June 1, 2008; Nice, France, Abstract F85". Journal of Bone & Joint Surgery, British Volume 92–B (SUPP II): 329.
- Turpie AG, Lassen MR, Davidson BL et al. (May 2009). "Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial".Lancet 373 (9676): 1673–80. doi:10.1016/S0140-6736(09)60734-0. PMID 19411100.
- ClinicalTrials.gov. "Randomized, Double-Blind Study Comparing Once Daily Oral Rivaroxaban With Adjusted-Dose Oral Warfarin for the Prevention of Stroke in Subjects With Non-Valvular Atrial Fibrillation". Retrieved 2009-02-11.
- ClinicalTrials.gov. "MAGELLAN - Multicenter, Randomized, Parallel Group Efficacy Superiority Study in Hospitalized Medically Ill Patients Comparing Rivaroxaban with Enoxaparin". Retrieved 2009-02-11.
- ClinicalTrials.gov. "Once-Daily Oral Direct Factor Xa Inhibitor Rivaroxaban in the Long-Term Prevention of Recurrent Symptomatic Venous Thromboembolism in Patients With Symptomatic Deep-Vein Thrombosis or Pulmonary Embolism. The Einstein-Extension Study". Retrieved 2009-02-11.
- ClinicalTrials.gov. "Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Deep-Vein Thrombosis (DVT) Without Symptomatic Pulmonary Embolism: Einstein-DVT Evaluation". Retrieved 2009-02-11.
- ClinicalTrials.gov. "Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Pulmonary Embolism (PE) With Or Without Symptomatic Deep-Vein Thrombosis: Einstein-PE Evaluation". Retrieved 2009-02-11.
- ClinicalTrials.gov. "A Randomized, Double-Blind, Placebo-Controlled, Event-Driven Multicenter Study to Evaluate the Efficacy and Safety of Rivaroxaban in Subjects With a Recent Acute Coronary Syndrome". Retrieved 2009-02-11.
- "Venous Thromboembolic Event (VTE) Prophylaxis in Medically Ill Patients (MAGELLAN)". ClinicalTrials.gov. 11 March 2011. Retrieved 15 April 2011.
- Hughes, Sue (5 April 2011). "MAGELLAN: Rivaroxaban prevents VTE in medical patients, but bleeding an issue". theheart.org. Retrieved 15 April 2011.
- "About the MAGELLAN Study". Bayer HealthCare. Retrieved 15 April 2011.
- Bauersachs, M.D., Rupert; The EINSTEIN Investigators (December 23, 2010). "Oral Rivaroxaban for Symptomatic Venous Thromboembolism". The New England Journal of Medecine 363 (26): 2499–2510. doi:10.1056/NEJMoa1007903. PMID 21128814. Retrieved 4 April 2011.
- "Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Deep-Vein Thrombosis Without Symptomatic Pulmonary Embolism: Einstein-DVT Evaluation". clinicaltrials.gov. Retrieved 15 April 2011.
- European Medicines Agency (2008). "CHP Assessment Report for Xarelto (EMEA/543519/2008)" (PDF). Retrieved 2009-06-11.
- Turpie AG (January 2008). "New oral anticoagulants in atrial fibrillation". European Heart Journal 29 (2): 155–65. doi:10.1093/eurheartj/ehm575. PMID 18096568.
| WO2013120465A1 * | Feb 18, 2013 | Aug 22, 2013 | Zentiva, K.S. | A process for the preparation of rivaroxaban based on the use of (s)-epichlorohydrin |
| WO2001047919A1 | Dec 11, 2000 | Jul 5, 2001 | Bayer Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| WO2004060887A1 | Dec 24, 2003 | Jul 22, 2004 | Bayer Healthcare Ag | Method for producing 5-chloro-n-({5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}-methyl)-2-thiophene carboxamide |
| WO2007116284A1 | Mar 26, 2007 | Oct 18, 2007 | Pfizer Prod Inc | Process for preparing linezolid |
| WO2009023233A1 | Aug 14, 2008 | Feb 19, 2009 | Concert Pharmaceuticals Inc | Substituted oxazolidinone derivatives |
| WO2010043110A1 | Oct 9, 2009 | Apr 22, 2010 | Changzhou Multiple Dimension Institute Of Industry Technology Co., Ltd. | A preparation method of high-purity l-carnitine |
| WO2010082627A1 | Jan 15, 2010 | Jul 22, 2010 | Daiso Co., Ltd. | Process for producing 2-hydroxymethylmorpholine salt |
| WO2010124835A1 | Apr 27, 2010 | Nov 4, 2010 | Belte Ag | Aluminium-silicon diecasting alloy for thin-walled structural components |
| WO2011080341A1 | Jan 3, 2011 | Jul 7, 2011 | Enantia, S.L. | Process for the preparation of rivaroxaban and intermediates thereof |
| WO2011098501A1 | Feb 10, 2011 | Aug 18, 2011 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2011102640A2 | Feb 16, 2011 | Aug 25, 2011 | Hanmi Holdings Co., Ltd. | Method for preparing sitagliptin and amine salt intermediates used therein |
| WO2012159992A1 * | May 18, 2012 | Nov 29, 2012 | Interquim, S.A. | Process for obtaining rivaroxaban and intermediate thereof |
| CN102786516A * | Aug 21, 2012 | Nov 21, 2012 | 湖南师范大学 | Method for synthesizing rivaroxaban |
| US7157456 | Dec 11, 2000 | Jan 2, 2007 | Bayer Healthcare Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| US7816355 * | Apr 28, 2009 | Oct 19, 2010 | Apotex Pharmachem Inc | Processes for the preparation of rivaroxaban and intermediates thereof |
| US20110034465 | Feb 10, 2011 | Apotex Pharmachem Inc. | Processes for the preparation of rivaroxaban and intermediates thereof |
| CN101560209A * | Apr 15, 2008 | Oct 21, 2009 | Shenyang, one hundred million Leo Pharmaceutical Co., Ltd. | Pyrimidine oxazolidinone compound and preparation method comprising |
| CN101619061A * | Aug 11, 2009 | Jan 6, 2010 | Shenyang Pharmaceutical University | Cyanopyridyl substituted oxazolidinone compounds |
| CN101821260A * | Aug 14, 2008 | Sep 1, 2010 | Consett Pharmaceuticals Ltd. | Substituted oxazolidinone derivatives |
| CN102250076A * | May 27, 2011 | Nov 23, 2011 | Hengdian Group homes Chemical Co., Ltd. | One kind of rivaroxaban Rivaroxaban intermediates and preparation methods |
| CN102250077A * | Jun 15, 2011 | Nov 23, 2011 | Zhejiang University | A method for intermediate and rivaroxaban Rivaroxaban for the synthesis of |
| CN102311400A * | Jun 29, 2010 | Jan 11, 2012 | Xiang really Biotechnology Co., Ltd. | Aminomethyl-3-aryl-2-oxazolidinone class method - Preparation of L-5- |
| CN102320988A * | Jun 3, 2011 | Jan 18, 2012 | Shanghai Institute of Organic Chemistry | 4- (4-aminophenyl) -3-morpholinone intermediate amide, the synthesis method and uses |
| EP2354128A1 * | Feb 10, 2010 | Aug 10, 2011 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2010124385A1 * | Apr 28, 2010 | Nov 4, 2010 | Apotex Pharmachem Inc. | Processes for the preparation of rivaroxaban and intermediates thereof |
FROM THE NET
RIVAROXABAN 5-Chloro-N-{[(5S) 2-oxo-3 [4-(3-oxo-4 ...
https://plus.google.com/.../posts/EeEveU8xvUd
32 mins ago - RIVAROXABAN 5-Chloro-N-{[(5S) 2-oxo-3 [4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide (Rivaroxaban) (1):1 rivaroxaban 1 ...WO 2015104605.new patent on Rivaroxaban, Wockhardt ...
https://plus.google.com/.../posts/g4TXSPBA4YV
1 hour ago - WO 2015104605.new patent on Rivaroxaban, Wockhardt Ltd Process for preparing rivaroxaban - comprising the reaction of a thioester compound and its salts ... | |
 | |
| Systematic (IUPAC) name | |
|---|---|
(S)-5-chloro-N-{[2-oxo-3-[4-(3-oxomorpholin-4-yl)
phenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide | |
| Clinical data | |
| Trade names | Xarelto |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category | |
| Legal status |
|
| Routes of administration | oral |
| Pharmacokinetic data | |
| Bioavailability | 80% to 100%; Cmax = 2 – 4 hours (10 mg oral)[1] |
| Metabolism | CYP3A4 , CYP2J2 and CYP-independent mechanisms[1] |
| Biological half-life | 5 – 9 hours in healthy subjects aged 20 to 45[1][2] |
| Excretion | 2/3 metabolized in liver and 1/3 eliminated unchanged[1] |
| Identifiers | |
| CAS Registry Number | 366789-02-8 |
| ATC code | B01AX06 |
| PubChem | CID: 6433119 |
| IUPHAR/BPS | 6388 |
| DrugBank | DB06228 |
| ChemSpider | 8051086 |
| UNII | 9NDF7JZ4M3 |
| ChEMBL | CHEMBL198362 |
| Synonyms | Xarelto, BAY 59-7939 |
| Chemical data | |
| Formula | C19H18ClN3O5S |
| Molecular mass | 435.882 g/mol |
 |
Each XARELTO tablet contains 10 mg, 15 mg, or 20 mg of rivaroxaban. The inactive ingredients of XARELTO are: croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. Additionally, the proprietary film coating mixture used for XARELTO 10 mg tablets is Opadry® Pink and for XARELTO 15 mg tablets is Opadry® Red, both containing ferric oxide red, hypromellose, polyethylene glycol 3350, and titanium dioxide, and for XARELTO 20 mg tablets is Opadry® II Dark Red, containing ferric oxide red, polyethylene glycol 3350, polyvinyl alcohol (partially hydrolyzed), talc, and titanium dioxide.
3
BETRIXABAN

N-(5-chloropyridin-2-yl)-2-([4-(N,N-dimethylcarbamimidoyl)benzoyl]amino)-5-methoxybenzamide
Betrixaban:PRT-54021, PRT-021, MK-4448, PRT-054021
N- (5- chloro-2-pyridyl) -2 – [[4 – [(dimethylamino) methyl] benzoyl] amino] -5 – methoxy – benzamideCAS 330942-05-7
MW 451.91, C23H22ClN5O3
Venous Thromboembolism (VTE)
Millennium INNOVATOR
Takeda Pharmaceutical Co Ltd
Lee’s Pharmaceutical Holdings (Hong Kong) Ltd; Portola Pharmaceuticals Inc…DEVELOPERS
PHASE 3 for Venous Thromboembolism (VTE)
Portola Pharmaceuticals, under license from Takeda (formerly known as Millennium Pharmaceuticals), is developing betrixaban (was reported to be in phase III in November 2015), for treating venous thrombosis
In October 2015, betrixaban was granted Fast Track designation by the FDA for extended-duration prevention of VTE or blood clots in acute medically ill patients
Betrixaban (INN, codenamed PRT-054,021) is an anticoagulant drug which acts as a direct factor Xa inhibitor.[1] It is potent, orally active and highly selective for factor Xa, being selected from a group of similar compounds for its low hERG affinity.[2] Betrixaban has undergone human clinical trials for prevention of embolism after knee surgery,[3] and prevention of stroke following atrial fibrillation,[4] with promising results.[5] Betrixaban is currently being studied in a human clinical trial for extended duration thromboprophylaxis to prevent venous thromboembolism in acute medically ill patients.[6] Joint development with Portola was discontinued in 2011 by Merck.[7] Betrixaban is now being developed by Portola Pharmaceuticals.
Long-acting, oral, direct Factor Xa Inhibitor
Description
Betrixaban is an oral small molecule anticoagulant that directly inhibits the activity of Factor Xa, an important validated target in the blood coagulation pathway.Key Characteristics
Betrixaban has been specifically designed for chronic, once-a-day treatment. It has a half-life that supports true, once-daily dosing and a low peak-to-trough drug concentration ratio that minimizes anticoagulant variability. Betrixaban is primarily eliminated unchanged in the bile and has been studied in patients with all degrees of renal function, including those with severe renal impairment (excluding dialysis patients). Betrixaban is minimally metabolized through the Cytochrome 450 enzyme system, which may result in low potential for CYP-related drug interactions. Betrixaban is reversible with PRT4445, a universal Factor Xa inhibitor antidote that Portola is developing as a companion product.Potential Indications
Treatment or prevention of life-threatening blood clots (venous thromboembolism; VTE) in acute medically ill patients.Clinical Development
ClinicalTrials.gov Identifier:
NCT01583218
COMPLETION-August 2014
http://clinicaltrials.gov/ct2/show/NCT01583218Portola has initiated its pivotal Phase 3 APEX Study to demonstrate the safety and efficacy of betrixaban for extended duration venous thromboembolism (VTE) prophylaxis for up to 35 days in acute medically ill patients with restricted mobility and certain risk factors. This randomized, double-blind, active-controlled, multicenter, multinational study will compare a once-daily dose of 80 mg of betrixaban for a total of 35 days (including both in the hospital and after discharge) with in-hospital administration of 40 mg of enoxaparin once daily for 6 to 14 days followed by placebo. The global study is expected to enroll approximately 6,850 patients at more than 400 study sites throughout the world. The primary objective of the trial is to demonstrate the superiority of betrixaban as compared to the current standard of care in the reduction of VTE-related events at 35 days while maintaining a favorable benefit to risk profile.
The APEX study is adequately powered to show a clinically relevant benefit with a p-value of less than 0.01 on the primary endpoint of total asymptomatic proximal DVT (as detected by ultrasound), symptomatic DVT (proximal or distal), non-fatal pulmonary embolism and VTE-related death. The first patient was enrolled in March 2012.
The safety and tolerability of betrixaban for stroke prevention was evaluated in 508 patients with atrial fibrillation in the Phase 2 EXPLORE-Xa dose-ranging study. Results were presented during a late-breaking session at the American College of Cardiology’s 59th Annual Scientific Session in March 2010. The data showed that a once-daily dose of oral betrixaban, given to patients with non-valvular atrial fibrillation or atrial flutter and at least one risk factor for stroke, reduced the incidence of major and clinically relevant non-major bleeds compared to dose-adjusted warfarin. In August 2010, additional pharmacodynamic data from a pre-specified analysis of EXPLORE-Xa showed a concentration dependent relationship and provided further evidence for the anticoagulant activity of betrixaban across all three doses studied in the clinical trial. The additional pharmacodynamic analysis provides information for dose selection for Phase 3 evaluation of betrixaban.
In 2007, positive top-line results from EXPERT were published in The Journal of Thrombosis and Haemostasis. This randomized, multi-center, Phase 2 in-hospital efficacy and safety study of the prevention of VTE compared betrixaban with enoxaparin in 215 patients undergoing knee replacement surgery.
Betrixaban (INN, codenamed PRT-054,021) is an anticoagulant drug which acts as a direct factor Xa inhibitor.[1] It is potent, orally active and highly selective for factor Xa, being selected from a group of similar compounds for its low hERG affinity.[2] Betrixaban has undergone human clinical trials for prevention of embolism after knee surgery,[3] and prevention of stroke following atrial fibrillation,[4] with promising results.[5]
Patent Document CN1391555A first discloses a preparation method (see Scheme 1):
CN101595092A (See Scheme 2).

Patent Document CN102762538A (see Scheme 3).
[0013]

CN104693114
Machine translated from chinese please bear with names
http://www.google.com/patents/CN104693114A?cl=en
Preparation Example 1 shell song in Spanish

Under stirring, temperature 15 ~ 20 ° C, the compound of formula II 50. 0g (0 123mol, leq.) Was mixed with 500ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition continued at 25 The reaction was stirred for ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 700ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water; the filter cake was mixed by stirring with 500ml of acetone, the pH adjusted with triethylamine to 7-8; filtered; the cake at 40 ~ 45 ° C and dried under reduced pressure to give Pui Spanish song 45. 5g. . Yield: 82 0%; HPLC purity: 98.9%, of which 0.05% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Take the above Tony Qu Spanish 45.0g, at about 70 ° C under stirring dissolved in N, N- dimethylacetamide 180ml, a toluene solution of 360ml; cooling crystallization, filtration, the filter cake washed with an appropriate amount of acetone at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish HPLC purity 99.7%.
(+) LC-MS: m / z = 452 ([M + H] +). Che NMR (400MHz, DMS0-d6) S:…. 2 96 (s, 6H), 3 83 (s, 3H), 7. 06-7 09 (dd, 1H), 7. 55-7 59 ( m, 3H), 7. 80-7. 83 (dd, 1H), 8. 21-8. 23 (d, 1H), 8. 27-8. 30 (d, 2H), 8. 37-8. 40 (d, 1H), 8. 41-8. 43 (d, 1H), 10. 54 (br., 2H).
Preparation Example 2 Tony Qu Spanish maleate
Under stirring, temperature 0 ~ 5 ° C, dropping 2mol isopropyl magnesium chloride in tetrahydrofuran / L (available commercial available) 105ml (0 • 21mol, 8. 4eq) twenty methylamine hydrochloride 8. 91g (0 • llmol, 4. 4eq) in tetrahydrofuran 60ml of the suspension, the reaction solution obtained dimethylamine.
Under stirring, temperature 0 ~ 5 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, and then dropping the above reaction liquid dimethylamine; After the addition continued 10 The reaction was stirred for ~ 15 ° c, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 10 ~ 15 ° C, the reaction solution was added to an aqueous solution of 45g and 100ml dubbed maleic acid solution; the organic solvent was evaporated under reduced pressure and concentrated, filtered concentrate precipitated solid cake was washed with the right amount of water washing. Cake at 40 ~ 45 ° C and dried under reduced pressure to give Tony Qu Spanish maleate 12.lg. . Yield: 85 4%; HPLC purity: 98.6%, which is 0.03% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Take the above shellfish Spanish song maleate 10. 0g, at about 70 ° C under stirring dissolved in a mixed solvent of ethanol 50ml and 25ml of water, dropping water 150ml; cooling crystallization, filtration, the filter cake at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish maleate HPLC purity 99.9%.
: HNMR (400MHz, DMS〇-d6) 8: 3. 25 (s, 3H), 3. 32 (s, 3H), 3. 87 (s, 3H), 6. 02 (s, 2H) , 7. 19-7. 21 (dd, 1H), 7. 44-7. 45 (1H), 7. 75-7. 77 (d, 2H), 7. 97-9. 98 (d, 2H) , 8. 08-8. 13 (m, 3H), 8. 44-8. 45 (d, 1H), 9. 01 (br., 1H), 9. 37 (br., 1H), 11.04 (s , 1H), 11. 13 (s, 1H).
Preparation Example 3 Tony Spanish song of [0075] Example
Under stirring, temperature 25 ~ 30 ° C, isopropylmagnesium chloride in tetrahydrofuran was added dropwise a solution of 2mol / L (available commercially available) 81ml (0 • 161mol, 7eq) to 2mol / L dimethylamine THF Solution (commercially available can) 121ml (0 • 242mol, 10. 5eq) to give dimethylamine reaction solution.
Under stirring, temperature 25 ~ 30 ° C, the hydrochloride salt of a compound of formula II 10. 0g (0 023mol, leq.) Was mixed with 100ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition was complete The reaction continued stirring at 25 ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 210ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water; the filter cake with 90ml acetone was stirred and mixed, the pH adjusted with triethylamine to 7-8; filtration; cake was 45 ~ 50 ° C and dried under reduced pressure to give Pui Qu Spanish 8. 35g. Yield: 80.5%. HPLC purity: 98.7%, which is 0.03% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Preparation Example 4 shellfish Spanish song hydrochloride
Under stirring, temperature 15 ~ 20 ° C, dropping lmol / n-amyl magnesium bromide tetrahydrofuran solution (which can be commercialized available) 75ml (0 • 075mol, 3eq) to 2mol / L of dimethyl L amine in tetrahydrofuran (commercially available can) 56ml (0 • 113mol, 4. 5eq) to give dimethylamine reaction solution.
Under stirring, temperature 15 ~ 20 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, was added dropwise the above-described reaction solution of dimethylamine; After the addition continued at 25 The reaction was stirred for ~ 30 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, at 15 ~ 20 ° C, the reaction solution was added to about 2mol L hydrochloric acid solution 100ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake washed with an appropriate amount of water. Cake at 40 ~ 45 ° C and dried under reduced pressure to give Tony Qu Spanish hydrochloride 10.lg, yield:. 82 9%; HPLC purity: 99.0%, which is 0.02% dechlorination impurity VIII, from A impurities IX was not detected.
Take the above shellfish Spanish song hydrochloride 10. 0g, at about 70 ° C under stirring dissolved in N, N- dimethylacetamide 40ml, a toluene solution of 80ml; cooling crystallization, filtration, cake at 40 ~ 45 ° C and dried under reduced pressure; the resulting song Tony Spanish hydrochloride HPLC purity 99.8%.
Preparation 5 shellfish Spanish song of [0082] Example
Under stirring, temperature 0 ~ 5 ° C, dropping lmol / diethyl zinc toluene solution of L (available commercially oriented) 50ml (0. 050mol, 2eq) to 2mol / L dimethylamine tetrahydrofuran (commercially available can) 28ml (0. 055mol, 2. 2eq) to give dimethylamine reaction solution.
Under stirring, temperature 0 ~ 5 ° C, the compound of formula II 10. 0g (0 025mol, leq.) Was mixed with 100ml of tetrahydrofuran, and then dropping the above reaction liquid dimethylamine; After dropping 5 continues The reaction was stirred for ~ 10 ° C, the progress of the reaction was monitored by HPLC. After completion of the reaction, in the next 5 ~ 10 ° C, the reaction mixture was added to about 2mol L dilute hydrochloric acid solution 70ml / in hydrochloric acid and then adjusting the pH to 2-3; concentrated under reduced pressure and the organic solvent was evaporated, filtered and concentrated liquid The precipitated solid, the filter cake was washed successively with a suitable amount of water; the filter cake with acetone l〇〇ml mixing, the pH adjusted with triethylamine to 7-8; filtered; the cake at 40 ~ 45 ° C under reduced pressed and dried to give Tony Qu Spanish 9. 03g. . Yield: 80 1%; HPLC purity: 99.0%, which is 0.02% dechlorinated impurities VIII, IX desmethyl impurities were not detected.
Preparation of compounds of Formula II Preparation Example 1
Methoxy-2-nitro – (5-chloro-pyridin-2-yl) -5 – benzamide (compound V) Preparation of [0086] (1) N-

(2) 2-Amino -N- (5- chloro – pyridin-2-yl) -5-methoxy – benzamide (compound IV) is prepared

(3) N- (5- chloro – pyridin-2-yl) -2- (4-cyano – benzoyl – amino) -5-methoxy – benzamide (compound II) is prepared

(+) LC-MS: m / z = 407 ([M + H] +). Insect NMR (400MHz, DMS0-d6) S:… 3 85 (s, 3H), 7 16-7 .19 (dd, 1H), 7. 39-7 41 (d, 1H), 7. 93- 7. 96 (d, 2H), 8. 02-8. 04 (m, 4H), 8. 13-8. 14 (d, 2H), 8. 42-8. 43 (d, 1H), 11. 06 (br. 2H).
Example 2 Preparation of the hydrochloride salt of the compound of formula II
at 10 ~ 20 ° C, a solution of a compound of formula IV 40. 0g (0 • 14mol, 1.Oeq) was dissolved in 400ml of tetrahydrofuran, a solution of cyanobenzoyl chloride (Compound III, can be commercialized available) 24 8g (0 15mol, 1.leq) and tetrahydrofuran solution 200ml dubbed, HPLC monitoring progress of the reaction;.. After the reaction was filtered, the filter cake washed with ethanol and after an appropriate amount, and dried under reduced pressure to obtain a compound of formula II hydrochloride . HPLC purity: 99.5%.
WO 2015176591
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015176591&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=FullTextExample 1: Preparation of Spanish Preparation and Form A half-L- malic acid shellfish song
At 55 ~ 60 ℃, the shellfish song Spanish 6.0g (13.3mmol), L- malic acid 1.1g (8.0mmol) was dissolved in tetrahydrofuran 70mL / water 7mL mixed solvent acetone was added with stirring 60mL, cooled to room temperature, Crystallization. Precipitated solid was filtered, and the resulting solid at 40 ~ 45 ℃ vacuum dried to give half L- malic acid shellfish Spanish song.
1H NMR(400MHz,MeOD)δ:2.355-2.419(dd,0.5H),2.735-2.781(dd,0.5H),3.226(s,6H),3.907(s,3H),4.302-4.326(dd,0.5H),7.195-7.224(dd,1H),7.448-7.455(d,1H),7.744-7.764(d,2H),7.821-7.849(dd,1H),8.145-8.165(d,2H),8.196-8.219(d,1H),8.238-8.261(d,1H),8.323-8.329(d,1H)。
Above 1 H-NMR results, δ: 3.907 (s, 3H) attributed to shellfish Spanish song molecule methyl CH 3 , 4.302-4.326 (dd, 0.5H) attributed to L- malic acid molecule methine CH , you can determine the song title product in shellfish Spanish and L- malic acid molar ratio of 2: 1.
PATENT
http://www.google.com.na/patents/EP2101760A2?cl=en
Example 2Preparation of the compound of Formula II

a. Gram scale preparation A slurry of the compound of Formula F (455 g, 1.0 eq.) in THF (4.67 kg,
10.3 parts) was prepared and adjusted to <10 0C. Lithium dimethyl amide was prepared as follows :hexyllithium (2.3 N/hexane, 2.45 L, 5.5 eq.) was added to dimethylamine solution (2 N/THF, 2.8 L, 5.5 eq.) maintaining <10 0C. The lithium dimethyl amide solution was charged into the slurry containing the compound of Formula F keeping the pot temperature of <10 0C. The reaction progress was monitored by in-process HPLC which confirmed that the amount of Formula F was <1.0 A%. A buffer solution of NaHCO3 (490 g, 1.1 parts, 5.7 eq.) and Na2CO3 (490 g, 1.1 parts, 4.5 eq.) in deionized water (6.6 kg, 14.51 parts) was prepared, and the above reaction mixture was transferred to this aqueous solution maintaining < 5 0C. The product precipitated out and the resulting slurry was adjusted to 20 0C over a period of 12 hr. The solid was filtered, and the resulting wet cake was washed with 3.5 kg (7.7 parts) of deionized water. The solid was filtered off using a coarse frit glass bench filter, and rinsed forwarded with cold (0-5 0C) absolute ethanol (628 g, 1.4 parts). The product was dried at 30-35 0C. Dry product was obtained in 458 g (73% yield). b. Kilogram scale preparation A slurry of the compound of Formula F (31.5 kg, 1.0 eq.) in THF (251 kg,8.0 parts) was prepared in a 780 L Hastelloy reactor (Reactor A) and adjusted to 0 0C (-3 to 3 0C). 2 M Dimethylamine in THF (161.0 kg, 5.0 eq.) and THF (63 kg, 2 parts) were charged into a 1900 L GLMS reactor (Reactor B) and adjusted to 0 0C (-3 to 3 0C) with maximum agitation. Hexyllithium (2.3 M, 97.2 kg, 4.5 eq.) was slowly charged to Reactor B while maintaining a max temperature of 10 0C. The pump and lines were rinsed forward to Reactor B with THF (3.2 kg). The Reactor B contents were adjusted to 0 0C (-3 to 3 0C), then transferred to Reactor A while keeping Reactor A temperature < 10 0C. The Reactor B pump and lines were rinsed forward with THF (31.4 kg, 1.0 part). The Reactor A contents were adjusted to 0 0C (-3 to 3 0C), and agitated at this temperature until the reaction was complete as verified by HPLC (1-2 hrs). After about 1 hr of agitation, in-process HPLC analysis indicated that 0 A% starting material remained (in-process criteria: max 1 A%). Reactor A contents were adjusted to -5 0C (-8 to -3 0C). In-process cleaning of Reactor B with water was performed. Two previously prepared aqueous solutions (NaHCO3 (35.0 kg, 1.1 parts) in water (236 kg, 7.5 parts), and Na2CO3 (35.0 kg 1.1 parts) in water (236 kg, 7.5 parts))were charged to Reactor B and adjusted to -3 0C (0 to 6 0C). Reactor A contents were transferred to Reactor B through an insulated line, maintaining the temperature of Reactor B at -8 0C to a maximum of 5 0C. The Reactor A pump and lines were rinsed forward with cold [-5 0C (-8 to -3 0C)] THF (31.4 kg, 1.0 part). Reactor B contents were adjusted to 22 0C (19-25 0C) and agitated for ca. 3 hrs. Slurry formation was visually confirmed, and Reactor B contents were filtered onto a 30″ centrifuge fitted with a filter cloth. The Reactor B pump and lines were rinsed forward onto the 30″ centrifuge fitted with a filter cloth with drinking water (63 kg, 2 parts). The wet filter cake (66.5 kg) was transferred back to Reactor B and submitted to a slurry wash in drinking water (1005 kg, 32 parts) at 22 0C (19-25) 0C for ca. 1 hr. The product was filtered onto the 30″ centrifuge (after in-process cleaning and fitting with a filter cloth), and the Reactor B lines and pump were rinsed forward with drinking water (63 kg, 2 parts). The water rinse was sampled for test by TDS, which was found to be 0.46%. The Reactor B pump, lines and wet filter cake were further rinsed with cold [0 0C (-3 to 3 0C)] ethanol (44 kg, 1.39 parts). The wet filter cake was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 35 0C. In-process LOD was 0% after ca. 24 hrs of drying, and the product was discharged (24.8 kg) in 76.7% yield. HPLC showed 98 % purity, with dechlorinated impurity at 1.14 %. Example 3
Preparation of the compound of Formula F Step 1. Synthesis of 2-nitro-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (C)

5-Methoxy-2-nitrobenzoic acid (A) (25.0 kg, 1.0 eq.), 2-amino-5- chloropyridine (B) (16.3 kg, 1.0 eq.), and acetonitrile (87.5 kg, 3.5 parts) were charged to a 380 L GLMS reactor. The reaction mixture was adjusted to 22 0C (19-25 0C) and anhydrous pyridine (30.0 kg, 3.0 eq.) was added. The pump and lines were rinsed forward with acetonitrile (22.5 kg, 0.9 parts), and the reactor contents were adjusted to a temperature of 19-22 0C. Phosphorous oxychloride (23.3 kg, 1.20 eq.) was charged to the contents of the reactor via a metering pump, while maintaining a temperature of 25 0C (22-28 0C). The metering pump and lines were rinsed forward with acetonitrile (12.5 kg, 0.5 parts), while keeping the temperature at 25 0C (22-28 0C). The reaction mixture normally turned from a slurry to a clear solution after the addition of about 1/3 of the POCI3. At the end of the addition, it became turbid. After complete addition, the reaction mixture was agitated at 25 0C (22-28 0C) for ca. 1 hr, at which time HPLC analysis confirmed reaction completion. The solution was cooled to 15 0C (12-18 0C) and drinking water (156.3 kg, 6.25 parts) was charged slowly while keeping reaction temperature of between 12 and 30 0C. The reaction mixture was then adjusted to 22 0C (19-25 0C) and agitated for ca. 5 hrs until exotherm ceased. Formation of a slurry was visually confirmed and the contents of the reactor were filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were washed forward onto the pressure nutsche with two portions of drinking water (62.5 kg, 2.5 parts each). The filtrate had a pH value of 7. The product (41.8 kg) was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 50 0C. After ca. 12 hrs, in-process LOD analysis indicated a solvent content of 0.72%. The dry product (C) was discharged (34.4 kg) with 88.2% yield and 99.1 % purity by HPLC. Step 2. Synthesis of 2-amino-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (D)


(17.2 kg, 1.1 eq.) and THF (92 kg, 3.5 parts). Reactor contents were agitated at 22 0C (19- 25 0C) until all of the solids had dissolved. The resulting solution was transferred to a lower receiver and the reactor was rinsed forward with THF (26 kg, 1 part). Compound D (26.4 kg, 1 eq.), THF (396 kg, 15 parts) and pyridine (2.90 kg, 0.4 eq.) were charged to a clean reactor. The pump and lines were rinsed forward with THF (34 kg, 1.3 parts). Via a metering pump, the 4-cyanobenzoyl chloride/THF solution was charged to the reactor, keeping the temperature at < 30 0C and rinsing forward with THF (ca. 10 kg). The resulting yellow-colored slurry was agitated at 22 0C (19-25 0C) for ca 2 hrs. In-process HPLC taken after 2 hrs showed a compound of Formula D content of 0%, indicating completion of the reaction. The slurry was filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, lines and wet cake were rinsed with three portions of ethanol (ca. 15 kg each). The wet filter cake was discharged (65.4 kg) and transferred back to the reactor for slurry wash in ethanol (317 kg, 12 parts) at 22 0C (19-25 0C) for ca. 1 hr. The slurry was filtered onto the pressure nutsche and the reactor, pump, lines, and wet filter cake were rinsed with two portions of ethanol (ca. 15 kg each) and two portions of THF (ca. 15 kg each). The wet filter cake was dried under vacuum with a maximum temperature of warm glycol bath (to heat the dryer jacket) of 40 0C. After 14.5 hrs of drying, LOD was 0.75%. The dried material was milled (screen 0.125″) to give 31.8 kg of product, which was dried under vacuum for another 10.5 hrs. LOD after drying was 1.8%, and the product was discharged (31.5 kg) in 74.8% yield (expected 60-90%). HPLC showed 100 % purity.
PATENT
http://www.google.com/patents/WO2011084519A1?cl=enU.S. Patent No. 6,376,515 B2 discloses a class of benzamide based compounds as specific factor Xa inhibitors. In particular, U.S. Patent No. 6,376,515 B2 describes a compound identified as Example 206, which is also disclosed in U.S. Patent No. 6,835,739 B2 as Example 206 and herein identified as betrixaban, which has the chemical formula of Formula I:

Scheme 1

Example 1: Preparation of betrixaban
 [0113] Dimethylformamide (13L) and hydrochloride (18 mL) were charged
into a reactor. Compound B (1 kg) was added followed by Compound A
(0.88 kg).
[0113] Dimethylformamide (13L) and hydrochloride (18 mL) were charged
into a reactor. Compound B (1 kg) was added followed by Compound A
(0.88 kg).
Compound A is commercially available or, just as with Compound B may be prepared using the methods described in Examples 4 and 5. The reaction mixture was cooled between 0 °C and -10 °C. EDC (0.752 kg) was added while maintaining the temperature between -10 °C and 0 °C. The reaction mixture was stirred until the content of
Compound B is below 0.10% area by HPLC. The reaction mixture was stirred until betrixaban started to crystallize. Acetone (26 L) was then added during a period of at least 1 hr while the temperature was maintained at between -10 °C and 0 °C. The suspension was then stirred for additional 2 hrs at a temperature of between 0 °C and 10 °C. The suspension was filtered and washed with cold acetone to give a wet product betrixaban. Example 2: Preparation of a maleate salt of betrixaban
 [0114] The wet betrixaban obtained above was reacted with maleic acid
(0.52 x weight of maleic acid/weight of dry betrixaban) in ethanol
(22.4 x volume of
[0114] The wet betrixaban obtained above was reacted with maleic acid
(0.52 x weight of maleic acid/weight of dry betrixaban) in ethanol
(22.4 x volume of
liquid/weight of dry betrixaban (v/w)) and purified water (5.7 x v/w) to form a betrixaban maleate salt. The solution of the betrixaban maleate salt was filtered and concentrated under vacuum until a final volume of 5.7 x v/w. Water (2 x v/w) was then added and the mixture was back concentrated until the same volume. The procedure of adding water and distil until a final volume of 5.7 x v/w was carried out until the molar ratio between the content of ethanol and the content of betrixaban maleate salt in the mixture was lower than, or equal to, 6. Betrixaban maleate salt crystallized during the removal of ethanol. The suspension was cooled to a temperature between 19 °C and 25 °C and stirred for not less than 2 hours at this temperature range. Betrixaban maleate salt was isolated by filtration, washed with water and dried under vacuum at a maximum temperature of 40 °C until the content of water was lower than, or equal to, 0.5 % w/w by Karl-Fisher. The purity of the maleate salt was determined to be greater than 99 % by HPLC. The betrixaban maleate isolated was in a crystalline form A which was concluded based on IR, DSC and XRPD results obtained, see Figures 3-5, respectively. The major peaks of XRPD pattern of crystalline form A are also listed in Table 2. Table 2: Betrixaban Form A XRPD Peak °2-Theta (2Θ0)
 Example 3: Synthesis of 2-nitro-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (C)
Example 3: Synthesis of 2-nitro-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide (C)
 D E C
D E C
[0115] 5-Methoxy-2-nitrobenzoic acid (D) (25.0 kg, 1.0 eq.), 2-amino-5- chloropyridine (E) (16.3 kg, 1.0 eq.), and acetonitrile (87.5 kg) were charged to a 380 L glass-lined reactor. The reaction mixture was adjusted to 22 °C (19-25 °C) and anhydrous pyridine (30.0 kg, 3.0 eq.) was added. The pump and lines were rinsed forward with acetonitrile (22.5 kg), and the reactor contents were adjusted to a temperature of 19-22 °C. Phosphorous oxychloride (23.3 kg, 1.20 eq.) was charged to the contents of the reactor via a metering pump, while maintaining a temperature of 25 °C (22-28 °C). The metering pump and lines were rinsed forward with acetonitrile (12.5 kg), while keeping the temperature at 25 °C (22-28 °C). The reaction mixture normally turned from a slurry to a clear solution after the addition of about 1/3 of the POCI3. At the end of the addition, it became turbid. After complete addition, the reaction mixture was agitated at 25 °C (22- 28 °C) for ca. 1 hr, at which time HPLC analysis confirmed reaction completion. The solution was cooled to 15 °C (12-18 °C) and water (156.3 kg) was charged slowly while keeping reaction temperature of between 12 and 30 °C. The reaction mixture was then adjusted to 22 °C (19-25 °C) and agitated for ca. 5 hrs until exotherm ceased. Formation of a slurry was visually confirmed and the contents of the reactor were filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were washed forward onto the pressure nutsche with two portions of water (62.5 kg). The filtrate had a pH value of 7. The product (41.8 kg) was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. After ca. 12 hrs, in-process LOD analysis indicated a solvent content of 0.72%. The dry product (C) was discharged (34.4 kg) with 88.2% yield and 99.1 % purity by HPLC.
Exam le 4. Synthesis of 2-amino-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide
 Process A
Process A
[0116] To a 780 L Hastelloy reactor, Compound C (33 kg, 1.0 eq.), 5%> platinum carbon (sulfided, 0.33 kg) and dichloromethane (578 kg) were charged. Agitation was started and reactor contents were adjusted to 22 °C (19-25 °C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 28 °C (25-31 °C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 28 °C (25 to 31 °C; maximum 31 °C) until the reaction was complete by HPLC. After 16.5 hrs, the reaction was deemed complete after confirming the disappearance of starting material (0.472 A%). The contents of the reactor were circulated through a conditioned Celite™ (diatomaceous earth; Celite Co., Santa Barbara, Ca.) pad (0.2-0.5 kg Celite™ conditioned with 20-55 kg dichloromethane) prepared in a 8″ sparkler filter to remove the platinum catalyst. The reactor and Celite™ bed were rinsed forward with two portions of dichloromethane (83 kg). The filtrate was transferred to and concentrated in a 570 L glass-lined reactor under an atmospheric pressure to ca. 132 L. Ethanol (69 kg) was charged and concentration continued under atmospheric pressure to ca. 99 L. In-process NMR indicated that the dichloromethane content was 39%. Ethanol (69 kg) was charged again and concentration continued again to ca. 99 L. In-process NMR indicated that the dichloromethane content was 5%. The reaction mixture was then adjusted to 3 °C (0 to 6 °C), agitated for ca. 1 hr, and the resulting slurry filtered onto a jacketed pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were rinsed forward with cold [3 °C (0-6 °C)] ethanol (26 kg. The wet filter cake (36.6 kg) was dried under vacuum at 40-50 °C with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The dry product (B) was discharged (26.4 kg) in 89.5% yield. HPLC showed 98.4 A% purity, with dechlorinated impurity at 0.083 %.
Process B
[0117] To a 780 L Hastelloy reactor, Compound C (33 kg, 1.0 eq.), 5%> platinum carbon (sulfided, 0.33 kg) and dichloromethane (578 kg) were charged. Agitation was started and reactor contents were adjusted to 22 °C (19-25 °C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 26 °C (21 to 31 °C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 26 °C (21 to 31 °C; maximum 31 °C) until the reaction was complete by HPLC. After 16.5 hrs, the reaction was deemed complete after confirming the disappearance of starting material (0.472 A%). The contents of the reactor were circulated through a conditioned Celite™ pad (0.2-0.5 kg Celite™ conditioned with 20-55 kg dichloromethane) prepared in a 8″ sparkler filter to remove the platinum catalyst. The reactor and Celite™ bed were rinsed forward with two portions of dichloromethane (83 kg). The filtrate was transferred to and concentrated in a 570 L glass-lined reactor under vacuum and a maximum temperature of 45 °C to ca. 132 L. Ethanol (69 kg) was charged and concentration continued under vacuum and a maximum temperature of 45 °C to ca. 132 L. In-process NMR indicated that the dichloromethane content was 39%. Ethanol (69 kg) was charged again and concentration continued again to ca. 132 L. In-process NMR indicated that the dichloromethane content was 5%. The reaction mixture was then adjusted to 3 °C (0 to 6 °C), agitated for ca. 1 hr, and the resulting slurry filtered onto a jacketed pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were rinsed forward with cold [3 °C (0-6 °C)] ethanol (26 kg. The wet filter cake (36.6 kg) was dried under vacuum at 40-50 °C with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The dry product (B) was discharged (26.4 kg) in 89.5% yield. HPLC showed 98.4 A% purity, with dechlorinated impurity at 0.083 %.
Example 5. Synthesis of 4-(N,N-dimethylcarbamimidoyl)benzoic acid (A)
Process A
 Step 1: Amidine Formation
Step 1: Amidine Formation
[0118] To a tetrahydrofuran solution of 2M dimethylamine, 2.3M hexane solution of hexyllithium was slowly added over a period of at least three (3) hours while maintaining the temperature at between -8°C and -12°C. This solution was added to the tetrahydrofuran solution of ethyl-4-cyanobenzoate (F) while maintaining the temperature between -8°C and -12°C. The completion of the reaction was confirmed by HPLC, and the solution temperature was adjusted to between -8°C and 3°C. The reaction mixture was slowly added to the cold solution of aqueous sodium bicarbonate solution and the desired ethyl-4-(N,N-dimethylcarbamimidoyl)benzoate (G) was extracted with ethyl acetate. The ethyl acetate layer was dried, filtered and evaporated under vacuum to afford ethyl-4-(N,N-dimethylcarbamimidoyl)benzoate (G) as a white solid.
Step 2: Hydrolysis of ester
[0119] To a THF solution of ethyl -4(N,N-dimethylcarbamimidoyl)benzoate (G) was added an aqueous solution of lithium hydroxide (2 eq.) and the reaction mixture was stirred for 6 hr. The completion of the reaction was confirmed by HPLC. To the reaction mixture was added water, followed by extraction with ethyl acetate. The aqueous layer was acidified with 6N HCI to pH between 3-4 at which point the desired 4-(N,N- dimethylcarbamimidoyl)benzoic acid precipitated as the white solid. The white solid isolated was washed with hexane to afford 4-(N,N-dimethylcarbamimidoyl)benzoic acid as an hydrochloride salt (A).
Process B:
 Step 1: Ester Formation
Step 1: Ester Formation
[0120] To a methanolic solution of 4-cyanobenzoic acid was added concentrated sulfuric acid and refluxed the reaction for at least 12 hours. The completion of the reaction was confirmed by HPLC. The solution was cooled and the solvent was evaporated. To the residue was added ethyl acetate followed by washing with 10 % sodium hydroxide solution. The ethyl acetate layer was dried, filtered and evaporated to give desired 4-methyl cyanobenzoate as a white solid.
Step 2: Dimethylamidine formation
[0121] A stream of HCI (gas) was bubbled through a 0 °C solution of 4-methyl cyanobenzoate (1 mmol) in 50 mL of ethanol until saturation. The mixture was stirred at room temperature overnight and evaporated to afford compound P. The resulting residue was treated with dimethylamine hydrochloride (0.15 eq.) in 20 mL ethanol at reflux temperature for 4 hours. The solvent was removed at reduced pressure and the residue was washed with hexane to afford desired product Q as a light yellow solid.
Step 3: Ester hydrolysis
[0122] To a THF solution of ethyl-4(N,N-dimethylcarbamimidoyl)benzoate (Q) was added an aqueous solution of lithium hydroxide (2 eq.) and the reaction mixture was stirred for 6 hours. The completion of the reaction was confirmed by HPLC. To the reaction mixture was added water, followed by extraction with ethyl acetate. The aqueous layer was acidified with 6N HC1 to pH between 3-4 at which point the desired 4- (N,N-dimethylcarbamimidoyl)benzoic acid precipitated as the white solid. The white solid isolated was washed with hexane to afford 4-(N,N-dimethylcarbamimidoyl)benzoic acid as an hydrochloride salt (A).
Example 6: Preparation of betrixaban, free base
 [0123] To 100 mL round bottom flask, was added compound B (2.0 g,
obtained as in Example 4), compound A (1.98 g, obtained as in example
5), 20 mL N,N- dimethylacetamide. The reaction mixture was stirred
briefly so as to dissolve most of the solid, then con. HC1 (36
microliters) was added. To this thin slurry add EDC HCl (1.8 g total,
Aldrich) in 3 portions, 0.6 g each, 20 min apart. The reaction mixture
was stirred for 1.5 hours for complete reaction. [0124] To this reaction
was added 2.3 g sodium carbonate solution in 10 mL water while the
batch was cooled with water bath to keep the batch temperature 22-30 °C.
Vigorous agitation was required to keep the batch well mixed. Then 10
mL water was added. The batch was stirred at 22-25 °C for 30 min. After a
slurry was formed, 20 mL more water was added. The batch was stirred at
22 °C for 1 hour. The batch was filtered and the wet cake was washed
with 3×5 mL water, then 5 mL acetone. The cake was dried on the funnel
by suction. The weight of the dry cake is 2.95 g -2.92 g which is the
crude betrixaban. To purify the crude betrixaban obtained, 1.0 g of the
crude solid was mixed with 4 mL Ν,Ν-dimethylacetamide and heated to 70
°C for 30 min. Then add 8 mL toluene was added and the mixture was
heated for 30 min, then cooled to 22 °C over 1 h, then cooled to 0 °C,
aged at 0 °C for 2 hours, filtered, washed with 2×1 mL toluene. The cake
was dried on the funnel by suction to obtain 0.88 g pure betrixaban
(I).
[0123] To 100 mL round bottom flask, was added compound B (2.0 g,
obtained as in Example 4), compound A (1.98 g, obtained as in example
5), 20 mL N,N- dimethylacetamide. The reaction mixture was stirred
briefly so as to dissolve most of the solid, then con. HC1 (36
microliters) was added. To this thin slurry add EDC HCl (1.8 g total,
Aldrich) in 3 portions, 0.6 g each, 20 min apart. The reaction mixture
was stirred for 1.5 hours for complete reaction. [0124] To this reaction
was added 2.3 g sodium carbonate solution in 10 mL water while the
batch was cooled with water bath to keep the batch temperature 22-30 °C.
Vigorous agitation was required to keep the batch well mixed. Then 10
mL water was added. The batch was stirred at 22-25 °C for 30 min. After a
slurry was formed, 20 mL more water was added. The batch was stirred at
22 °C for 1 hour. The batch was filtered and the wet cake was washed
with 3×5 mL water, then 5 mL acetone. The cake was dried on the funnel
by suction. The weight of the dry cake is 2.95 g -2.92 g which is the
crude betrixaban. To purify the crude betrixaban obtained, 1.0 g of the
crude solid was mixed with 4 mL Ν,Ν-dimethylacetamide and heated to 70
°C for 30 min. Then add 8 mL toluene was added and the mixture was
heated for 30 min, then cooled to 22 °C over 1 h, then cooled to 0 °C,
aged at 0 °C for 2 hours, filtered, washed with 2×1 mL toluene. The cake
was dried on the funnel by suction to obtain 0.88 g pure betrixaban
(I).

Compound A is commercially available or, just as with Compound B may be prepared using the methods described in Examples 4 and 5. The reaction mixture was cooled between 0 °C and -10 °C. EDC (0.752 kg) was added while maintaining the temperature between -10 °C and 0 °C. The reaction mixture was stirred until the content of
Compound B is below 0.10% area by HPLC. The reaction mixture was stirred until betrixaban started to crystallize. Acetone (26 L) was then added during a period of at least 1 hr while the temperature was maintained at between -10 °C and 0 °C. The suspension was then stirred for additional 2 hrs at a temperature of between 0 °C and 10 °C. The suspension was filtered and washed with cold acetone to give a wet product betrixaban. Example 2: Preparation of a maleate salt of betrixaban

liquid/weight of dry betrixaban (v/w)) and purified water (5.7 x v/w) to form a betrixaban maleate salt. The solution of the betrixaban maleate salt was filtered and concentrated under vacuum until a final volume of 5.7 x v/w. Water (2 x v/w) was then added and the mixture was back concentrated until the same volume. The procedure of adding water and distil until a final volume of 5.7 x v/w was carried out until the molar ratio between the content of ethanol and the content of betrixaban maleate salt in the mixture was lower than, or equal to, 6. Betrixaban maleate salt crystallized during the removal of ethanol. The suspension was cooled to a temperature between 19 °C and 25 °C and stirred for not less than 2 hours at this temperature range. Betrixaban maleate salt was isolated by filtration, washed with water and dried under vacuum at a maximum temperature of 40 °C until the content of water was lower than, or equal to, 0.5 % w/w by Karl-Fisher. The purity of the maleate salt was determined to be greater than 99 % by HPLC. The betrixaban maleate isolated was in a crystalline form A which was concluded based on IR, DSC and XRPD results obtained, see Figures 3-5, respectively. The major peaks of XRPD pattern of crystalline form A are also listed in Table 2. Table 2: Betrixaban Form A XRPD Peak °2-Theta (2Θ0)


[0115] 5-Methoxy-2-nitrobenzoic acid (D) (25.0 kg, 1.0 eq.), 2-amino-5- chloropyridine (E) (16.3 kg, 1.0 eq.), and acetonitrile (87.5 kg) were charged to a 380 L glass-lined reactor. The reaction mixture was adjusted to 22 °C (19-25 °C) and anhydrous pyridine (30.0 kg, 3.0 eq.) was added. The pump and lines were rinsed forward with acetonitrile (22.5 kg), and the reactor contents were adjusted to a temperature of 19-22 °C. Phosphorous oxychloride (23.3 kg, 1.20 eq.) was charged to the contents of the reactor via a metering pump, while maintaining a temperature of 25 °C (22-28 °C). The metering pump and lines were rinsed forward with acetonitrile (12.5 kg), while keeping the temperature at 25 °C (22-28 °C). The reaction mixture normally turned from a slurry to a clear solution after the addition of about 1/3 of the POCI3. At the end of the addition, it became turbid. After complete addition, the reaction mixture was agitated at 25 °C (22- 28 °C) for ca. 1 hr, at which time HPLC analysis confirmed reaction completion. The solution was cooled to 15 °C (12-18 °C) and water (156.3 kg) was charged slowly while keeping reaction temperature of between 12 and 30 °C. The reaction mixture was then adjusted to 22 °C (19-25 °C) and agitated for ca. 5 hrs until exotherm ceased. Formation of a slurry was visually confirmed and the contents of the reactor were filtered onto a pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were washed forward onto the pressure nutsche with two portions of water (62.5 kg). The filtrate had a pH value of 7. The product (41.8 kg) was dried under vacuum with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. After ca. 12 hrs, in-process LOD analysis indicated a solvent content of 0.72%. The dry product (C) was discharged (34.4 kg) with 88.2% yield and 99.1 % purity by HPLC.
Exam le 4. Synthesis of 2-amino-N-(5-chloro-pyridin-2-yl)-5-methoxy-benzamide

[0116] To a 780 L Hastelloy reactor, Compound C (33 kg, 1.0 eq.), 5%> platinum carbon (sulfided, 0.33 kg) and dichloromethane (578 kg) were charged. Agitation was started and reactor contents were adjusted to 22 °C (19-25 °C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 28 °C (25-31 °C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 28 °C (25 to 31 °C; maximum 31 °C) until the reaction was complete by HPLC. After 16.5 hrs, the reaction was deemed complete after confirming the disappearance of starting material (0.472 A%). The contents of the reactor were circulated through a conditioned Celite™ (diatomaceous earth; Celite Co., Santa Barbara, Ca.) pad (0.2-0.5 kg Celite™ conditioned with 20-55 kg dichloromethane) prepared in a 8″ sparkler filter to remove the platinum catalyst. The reactor and Celite™ bed were rinsed forward with two portions of dichloromethane (83 kg). The filtrate was transferred to and concentrated in a 570 L glass-lined reactor under an atmospheric pressure to ca. 132 L. Ethanol (69 kg) was charged and concentration continued under atmospheric pressure to ca. 99 L. In-process NMR indicated that the dichloromethane content was 39%. Ethanol (69 kg) was charged again and concentration continued again to ca. 99 L. In-process NMR indicated that the dichloromethane content was 5%. The reaction mixture was then adjusted to 3 °C (0 to 6 °C), agitated for ca. 1 hr, and the resulting slurry filtered onto a jacketed pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were rinsed forward with cold [3 °C (0-6 °C)] ethanol (26 kg. The wet filter cake (36.6 kg) was dried under vacuum at 40-50 °C with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The dry product (B) was discharged (26.4 kg) in 89.5% yield. HPLC showed 98.4 A% purity, with dechlorinated impurity at 0.083 %.
Process B
[0117] To a 780 L Hastelloy reactor, Compound C (33 kg, 1.0 eq.), 5%> platinum carbon (sulfided, 0.33 kg) and dichloromethane (578 kg) were charged. Agitation was started and reactor contents were adjusted to 22 °C (19-25 °C). The reactor was pressurized with ca. 30 psi hydrogen and the reaction mixture gently heated to 26 °C (21 to 31 °C). Hydrogenation of the reactor contents was performed under ca. 30 psi at 26 °C (21 to 31 °C; maximum 31 °C) until the reaction was complete by HPLC. After 16.5 hrs, the reaction was deemed complete after confirming the disappearance of starting material (0.472 A%). The contents of the reactor were circulated through a conditioned Celite™ pad (0.2-0.5 kg Celite™ conditioned with 20-55 kg dichloromethane) prepared in a 8″ sparkler filter to remove the platinum catalyst. The reactor and Celite™ bed were rinsed forward with two portions of dichloromethane (83 kg). The filtrate was transferred to and concentrated in a 570 L glass-lined reactor under vacuum and a maximum temperature of 45 °C to ca. 132 L. Ethanol (69 kg) was charged and concentration continued under vacuum and a maximum temperature of 45 °C to ca. 132 L. In-process NMR indicated that the dichloromethane content was 39%. Ethanol (69 kg) was charged again and concentration continued again to ca. 132 L. In-process NMR indicated that the dichloromethane content was 5%. The reaction mixture was then adjusted to 3 °C (0 to 6 °C), agitated for ca. 1 hr, and the resulting slurry filtered onto a jacketed pressure nutsche fitted with a filter cloth. The reactor, pump, and lines were rinsed forward with cold [3 °C (0-6 °C)] ethanol (26 kg. The wet filter cake (36.6 kg) was dried under vacuum at 40-50 °C with a maximum temperature of water bath (to heat dryer jacket) of 50 °C. LOD analysis after 12.5 hrs indicated solvent content was at 0.1%. The dry product (B) was discharged (26.4 kg) in 89.5% yield. HPLC showed 98.4 A% purity, with dechlorinated impurity at 0.083 %.
Example 5. Synthesis of 4-(N,N-dimethylcarbamimidoyl)benzoic acid (A)
Process A

[0118] To a tetrahydrofuran solution of 2M dimethylamine, 2.3M hexane solution of hexyllithium was slowly added over a period of at least three (3) hours while maintaining the temperature at between -8°C and -12°C. This solution was added to the tetrahydrofuran solution of ethyl-4-cyanobenzoate (F) while maintaining the temperature between -8°C and -12°C. The completion of the reaction was confirmed by HPLC, and the solution temperature was adjusted to between -8°C and 3°C. The reaction mixture was slowly added to the cold solution of aqueous sodium bicarbonate solution and the desired ethyl-4-(N,N-dimethylcarbamimidoyl)benzoate (G) was extracted with ethyl acetate. The ethyl acetate layer was dried, filtered and evaporated under vacuum to afford ethyl-4-(N,N-dimethylcarbamimidoyl)benzoate (G) as a white solid.
Step 2: Hydrolysis of ester
[0119] To a THF solution of ethyl -4(N,N-dimethylcarbamimidoyl)benzoate (G) was added an aqueous solution of lithium hydroxide (2 eq.) and the reaction mixture was stirred for 6 hr. The completion of the reaction was confirmed by HPLC. To the reaction mixture was added water, followed by extraction with ethyl acetate. The aqueous layer was acidified with 6N HCI to pH between 3-4 at which point the desired 4-(N,N- dimethylcarbamimidoyl)benzoic acid precipitated as the white solid. The white solid isolated was washed with hexane to afford 4-(N,N-dimethylcarbamimidoyl)benzoic acid as an hydrochloride salt (A).
Process B:

[0120] To a methanolic solution of 4-cyanobenzoic acid was added concentrated sulfuric acid and refluxed the reaction for at least 12 hours. The completion of the reaction was confirmed by HPLC. The solution was cooled and the solvent was evaporated. To the residue was added ethyl acetate followed by washing with 10 % sodium hydroxide solution. The ethyl acetate layer was dried, filtered and evaporated to give desired 4-methyl cyanobenzoate as a white solid.
Step 2: Dimethylamidine formation
[0121] A stream of HCI (gas) was bubbled through a 0 °C solution of 4-methyl cyanobenzoate (1 mmol) in 50 mL of ethanol until saturation. The mixture was stirred at room temperature overnight and evaporated to afford compound P. The resulting residue was treated with dimethylamine hydrochloride (0.15 eq.) in 20 mL ethanol at reflux temperature for 4 hours. The solvent was removed at reduced pressure and the residue was washed with hexane to afford desired product Q as a light yellow solid.
Step 3: Ester hydrolysis
[0122] To a THF solution of ethyl-4(N,N-dimethylcarbamimidoyl)benzoate (Q) was added an aqueous solution of lithium hydroxide (2 eq.) and the reaction mixture was stirred for 6 hours. The completion of the reaction was confirmed by HPLC. To the reaction mixture was added water, followed by extraction with ethyl acetate. The aqueous layer was acidified with 6N HC1 to pH between 3-4 at which point the desired 4- (N,N-dimethylcarbamimidoyl)benzoic acid precipitated as the white solid. The white solid isolated was washed with hexane to afford 4-(N,N-dimethylcarbamimidoyl)benzoic acid as an hydrochloride salt (A).
Example 6: Preparation of betrixaban, free base

| WO2012031017A1 * | Aug 31, 2011 | Mar 8, 2012 | Merck Sharp & Dohme Corp. | CRYSTALLINE FORMS OF A FACTOR Xa INHIBITOR |
| WO2013033370A1 * | Aug 30, 2012 | Mar 7, 2013 | Portola Pharmaceuticals, Inc. | Prevention and treatment of thrombosis in medically ill patients |
| US8946269 | Aug 31, 2011 | Feb 3, 2015 | Portola Pharmaceuticals, Inc. | Crystalline forms of a factor Xa inhibitor |
| WO2004083174A2 * | Mar 17, 2004 | Sep 30, 2004 | Timur Gangor | Sulfonyl-amidino containing and tetrahydropyrimidino containing compounds as factor xa inhibitors |
| WO2008057972A1 | Nov 1, 2007 | May 15, 2008 | Millennium Pharm Inc | Methods of synthesizing pharmaceutical salts of a factor xa inhibitor |
| US6376515 | Feb 28, 2001 | Apr 23, 2002 | Cor Therapeutics, Inc. | Benzamides and related inhibitors of factor Xa |
| US6835739 | Oct 15, 2003 | Dec 28, 2004 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US6844367 | Sep 15, 2000 | Jan 18, 2005 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US61287680 |
References
- Eriksson BI, Quinlan DJ, Weitz JI (2009). “Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor xa inhibitors in development”. Clinical Pharmacokinetics48 (1): 1–22. PMID19071881.
- Zhang P, Huang W, Wang L, Bao L, Jia ZJ, Bauer SM, Goldman EA, Probst GD, Song Y, Su T, Fan J, Wu Y, Li W, Woolfrey J, Sinha U, Wong PW, Edwards ST, Arfsten AE, Clizbe LA, Kanter J, Pandey A, Park G, Hutchaleelaha A, Lambing JL, Hollenbach SJ, Scarborough RM, Zhu BY (April 2009). “Discovery of betrixaban (PRT054021), N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide, a highly potent, selective, and orally efficacious factor Xa inhibitor”. Bioorganic & Medicinal Chemistry Letters19 (8): 2179–85. doi:10.1016/j.bmcl.2009.02.111. PMID19297154.
- Turpie AG, Bauer KA, Davidson BL, Fisher WD, Gent M, Huo MH, Sinha U, Gretler DD (January 2009). “A randomized evaluation of betrixaban, an oral factor Xa inhibitor, for prevention of thromboembolic events after total knee replacement (EXPERT)”. Thrombosis and Haemostasis101 (1): 68–76. PMID19132191.
- Piccini, J. P.; Lopes, R. D.; Mahaffey, K. W. (2010). “Oral factor Xa inhibitors for the prevention of stroke in atrial fibrillation”. Current Opinion in Cardiology25 (4): 312. doi:10.1097/HCO.0b013e32833a524f. PMID20520539. edit
- Sobieraj-Teague, M.; O’donnell, M.; Eikelboom, J. (2009). “New Anticoagulants for Atrial Fibrillation”. Seminars in Thrombosis and Hemostasis35 (5): 515–24. doi:10.1055/s-0029-1234147. PMID19739042. edit
- 6 Cohen, Alexander T.; Harrington, Robert; Goldhaber, Samuel Z.; Hull, Russell; Gibson, C. Michael; Hernandez, Adrian F.; Kitt, Michael M.; Lorenz, Todd J. (2014-01-01). “The design and rationale for the Acute Medically Ill Venous Thromboembolism Prevention with Extended Duration Betrixaban (APEX) study”. American Heart Journal 167 (3). doi:10.1016/j.ahj.2013.11.006.
- 7
 |
|
| Systematic (IUPAC) name | |
|---|---|
N-(5-chloropyridin-2-yl)-2-([4-(N,N-dimethylcarbamimidoyl)benzoyl]amino)-5-methoxybenzamide
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Number | 330942-05-7 |
| ATC code | None |
| PubChem | CID: 10275777 |
| ChemSpider | 18981107 |
| UNII | 74RWP7W0J9 |
| ChEMBL | CHEMBL512351 |
| Chemical data | |
| Formula | C23H22ClN5O3 |
| Molecular mass | 451.905 g/mol |
/////////////CN(C)C(=N)C1=CC=C(C=C1)C(=O)NC2=C(C=C(C=C2)OC)C(=O)NC3=NC=C(C=C3)Cl
See...........http://newdrugapprovals.org/2013/03/05/phase-3-portola-pharma-betrixaban-long-acting-oral-direct-factor-xa-inhibitor/
4 EDOXABAN
Edoxaban, DU-176b
Edoxaban (DU-176b, trade names Savaysa, Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It was developed by Daiichi Sankyo and approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1] It was also approved by the FDA in January 2015 for the prevention of stroke and non–central-nervous-system systemic embolism.[2]
Daiichi Sankyo receives FDA approval for anti-clotting drug Savaysa
Japanese drug-maker Daiichi Sankyo has obtained approval from US Food and Drug Administration (FDA) for its anti-clotting drug Savaysa (edoxaban tablets)..8 JAN 2015
Daiichi Sankyo, APPROVED IN JAPAN as tosylate monohydrate salt in 2011 for the prevention of venous embolism in patients undergoing total hip replacement surgery
for synthesis see….http://www.sciencedirect.com/science/article/pii/S0968089613002642 Bioorganic & Medicinal Chemistry 21 (2013) 2795–2825, see s[pecific page 2808 for description ie 14/31 of pdf
WO 2010071121, http://www.google.com/patents/WO2010071121A1
WO 2007032498
N’-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide
- N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide
Edoxaban (INN, codenamed DU-176b, trade name Lixiana) is an anticoagulant drug which acts as a direct factor Xa inhibitor. It is being developed by Daiichi Sankyo. It was approved in July 2011 in Japan for prevention of venous thromboembolisms (VTE) following lower-limb orthopedic surgery.[1]

Daichi Sankyo’s edoxaban tosilate is an orally administered
coagulation factor Xa inhibitor that was approved and launched
in Japan for the preventive treatment of venous thromboembolic
events (VTE) in patients undergoing total knee arthroplasty, total
hip arthroplasty, or hip fracture surgery. Edoxaban has been
shown to have a rapid onset of anticoagulant effect due to short
Tmax (1–2 h) after dosing and sustained for up to 24 h post-dose.
Marketed under the brand name Lixiana, it is currently in phase
III studies in the US for the prevention of stroke and systemic embolic
events in patients with atrial fibrillation (AF) and venous
thromboembolism (VTE).
Several Phase II clinical trials have been conducted, for example for thromboprophylaxis after total hip replacement[3] (phase III early results compare well to enoxaparin[4]), and for stroke prevention in patients with atrial fibrillation[5][6].Those papers follow similar recent major trials showing similar results for the other new factor Xa inhibitors, rivaroxaban and apixaban.
A large phase III trial showed that edoxaban was non inferior to warfarin in preventing recurrent venous thromboembolic events with fewer episodes of major bleeding.[7]

.................
PATENThttp://www.google.com/patents/WO2014081047A1?cl=en
Chemically, edoxaban is
N1- (5-chloropyridin-2-yl) -N2- ( (IS, 2R/4S) -4- [ (dimethylamino) carbo nyl] -2- { [ ( 5-methyl-4 , 5,6, 7-tetrahydrothiazolo [5 , 4-c] pyridin-2-yl ) carbonyl] amino}eyelohexyl) ethanediamide , represented by the following formula (A) :


Edoxaban is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (also referred to as activated factor X or FXa) , and is useful as a preventive and/or therapeutic drug for thrombotic diseases.
Several processes are known in the literature for preparing edoxaban for example, U.S. Patent No. 7365205; U.S. Publication No . 20090105491.
U.S. Patent No. 7365205 provides a process for the preparation of edoxaban, wherein the process involves the use of
(IS, 4S, 5S) -4-iodo-6-oxabicyclo [3.2.1] octan-7-one, represented by the following formula (C) :

as an intermediate.
The present inventors have identified that
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one, represented by the following formula (I) :

could also be used as an intermediate for the preparation of FXa inhibitory compounds like edoxaban. The present inventors have found that replacement of
(IS, 4S, 5S) -4-iodo-6-oxabicyclo [3.2.1] octan-7-one (C) with
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one (I) has a better atom economy and also an impact on cost.
A method for the synthesis of the
(IS, 4S, 5S) -4 -bromo- 6 -oxabicyclo [3.2.1] octan-7-one (I) was reported in Tetrahedron Letters, 51, (2010) Pages 3433-3435 which involves the reaction of ( IS) -cyclohex-3 -ene- 1-carboxylic acid represented by the following formula (II) :

with N-bromosuccinimide in the presence of molecular sieves using dichloromethane as a solvent. However, this reaction is carried out in dark over a period of 7 hours and does not provide a pure product .
Tetrahedron, Vol. 28, Pages 3393 -3399 , 1972 provides a process for the preparation of 4 -bromo- 6 -oxabicyclo [3.2.1] octan-7-one which involves the addition of 20% excess of a 2M solution of bromine in chloroform to a stirred solution of cyclohex- 3 -ene- 1-carboxylic acid (0.04 mol) in chloroform (250 mL) in the absence of a base . Extraction with aqueous sodium bicarbonate followed by acidification gave, after extraction with ether and evaporation of the extract, a mixture of cis & trans 3 , 4-dibromocyclohexanecarboxylic acid (6.7 g) and evaporation of the chloroform layer afforded the bromolactone (0.59 g) . It further provides a process for the preparation of
4 -bromo-6 -oxabicyclo [3.2.1] octan-7-one which involves the treating of cyclohex-3-ene-l-carboxylic acid (0.08 mol) dissolved in chloroform (450 mL) with 20% excess bromine in the presence of an equimolar amount of triethylamine (8.1 g) . After extraction of the amine with 2N hydrochloric acid, and work-up, bromolactone (10.7 g) and a mixture of cis & trans 3 , 4 -dibromocyclohexanecarboxylic acid (6.6 g) were obtained.
Tetrahedron Vol. 48, No. 3, Pages 539-544, 1992 provides a process for the preparation of
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one (I) which involves the addition of 1M solution of bromine in chloroform (30 mL) at 0°C to a solution of ( IS) -cyclohex-3 -ene- 1-carboxylic acid (0.024 mol) of formula (II) in chloroform (600 mL) in the presence of an equimolar amount of triethylamine (3.33 mL) . After work-up, the crude bromolactone obtained was recrystallized from petroleum ether.
However, bromination using bromine does not provide a pure product in good yield.
Heterocycles, Vol. 23, No. 8, Pages 2035-2039, 1985 provides a process for the 4-bromo-6-oxabicyclo [3.2.1] octan-7-one which involves the addition of cyclohex-3-ene-l-carboxylic acid (1.0 mM) in 1 , 2 -dimethoxyethane (2 mL) to a stirred solution of 90% Lead (IV) acetate (1.1 or 2.2 mM) in 1 , 2 -dimethoxyethane (4 mL) followed by the addition of Zinc bromide (2.2 mM) in 1 , 2 -dimethoxyethane (4 mL) and continuing the stirring for 10-30 minutes at 0°C . The reaction mixture was poured into a solution of ice-cold water (30 mL) and 10% hydrochloric acid (10 mL) , and extracted with ether (50 mL X 3) . The combined ether extract was washed successively with saturated sodium hydrogen carbonate solution (20 mL) , 10% sodium thiosulphate solution (5 mL) , and brine (10 mL) , and dried over sodium sulphate. Evaporation of the solvent gave crude lactone which were separated and purified (42% yield) . However, this reaction does not provide a pure product in good yield.
Heterocycles, Vol. 31, No. 6, Pages 987-991, 1990 provides a method for bromolactonization using a
dimethylsulfoxide-trimethylsilyl bromide-amine system. The bromolactonization is carried out for 10 to 72 hours using different solvents and triethylamine or diisopropylethyl amine as base. However, this process does not provide a product in high yield. Further the process afforded the cis isomer exclusively. Journal of the Chemical Society, Perkin Transactions 1:
Organic and Bio-Organic Chemistry (1972-1999) (1994) , (7) , Pages 847-851 provides a method for bromolactonization using a
dimethylsulfoxide-trimethylsilyl bromide-amine system. The bromolactonization is carried out for 12 hours using
dimethylsulfoxide and chloroform solvent system and triethylamine or diisopropylethyl amine as base. However, this process resulted in a low yield of about 55%. Citation List
Patent Literature
PTLl: U.S. Patent No. 7365205
PTL2: U.S. Publication No. 20090105491.
Non Patent Reference
NPLl: Feng Chen et al . , Tetrahedron Letters, 51, (2010) Pages 3433-3435.
NPL2 : G. Belluci et al . , Tetrahedron, Vol. 28, No. 13, Pages 3393-3399, 1972.
NPL3 : Marco Chini et al ., Tetrahedron Vol .48, No. 3, Pages 539-544 , 1992.
NPL4 : Y. Fujimoto et al . , Heterocycles , Vol. 23, No. 8, Pages 2035-2039, 1985.
NPL5: C. Iwata et al . , Heterocycles, Vol. 31, No. 6, Pages 987-991, 1990. -
NPL6 : K. Miyashita et al . , Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999) (1994) , (7) , Pages 847-851.
Summary of Invention
Technical Problem
It is an object of the present invention to solve the problems associated with the prior art, and to provide an improved and efficient method for the preparation of
(IS, 4S, 5S) -4-bromo-6-oxabicyclo [3.2.1] octan-7-one of formula (I).
Solution to Problem As a result of conducting diligent studies to attain the object, the present inventors have found that: surprisingly, the use of N-bromosuccinimide or bromohydantoin (representative is
1, 3-dibromo-5, 5-dimethylhydantoin) as brominating agent in the presence of a base selected from calcium oxide or calcium hydroxide, in specific mole ratios in a solvent selected from the group consisting of dichloromethane , toluene, tetrahydrofuran, ethyl acetate, hexanes, cyclopentyl methyl ether (CPME) or a mixture thereof can efficiently produce a pure
( IS , 4S , 5S) -4 -bromo- 6 -oxabicyclo [3.2.1] octan- 7 -one (I) in better yields. The process provides obvious benefits with respect to economics, convenience to operate at a commercial scale.
................................
SEE
http://www.google.co.ug/patents/US20090105491
...............................
PATENT
http://www.google.com/patents/EP2589590A1?cl=en
FREE BASE
- (Reference
Example 6)
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide
(X) (production method described in the pamphlet of International
Publication No. WO 2007/032498)
- Methanesulfonic acid (66 ml) was added to a suspension of tert-butyl [(1R,2,S,5S)-2-({[(5-chloropyridin-2-yl)amino](oxo)acetyl}amino)-5-(dimethylaminocarbonyl)cyclohexyl]carbamate (5) (95.1 g) in acetonitrile (1900 ml) at room temperature, and the mixture was stirred at this temperature for 2 hours. To the reaction solution, triethylamine (155 ml), 5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyrzdine-2-carboxylic acid hydrochloride (8) (52.5 g), 1-hydroxybenzotriazole (33.0 g), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (46.8 g) were added under ice cooling, and the mixture was stirred at room temperature for 16 hours. Triethylamine and water were added thereto, and the mixture was stirred for 1 hour under ice cooling. Then, crystals were collected by filtration to obtain the title compound (X) (103.2 g). 1H-NMR (CDCl3) δ : 1.60-1.98 (3H, m), 2.00-2.16 (3H, m), 2.52 (3H, s), 2.78-2.90 (3H, m), 2.92-2.98 (2H, m), 2.95 (3H, s), 3.06 (3H, s), 3.69 (1H, d, J = 15.4 Hz), 3.75 (1H, d, J = 15.4 Hz), 4.07-4.15 (1H, m), 4.66-4.72 (1H, m), 7.40 (1H, dd, J = 8.8, 0.6 Hz), 7. 68 (1H, dd, J = 8.8, 2.4 Hz), 8.03 (1H, d, J = 7.8 Hz), 8.16 (1H, dd, J = 8.8, 0.6 Hz), 8.30 (1H, dd, J = 2. 4, 0.6 Hz), 9.72 (1H, s). MS (ESI) m/z: 548 (M+H)+.

- (Reference
Example 7)
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide
mono-p-toluenesulfonate monohydrate (X-a) (production method described
in the pamphlet of International Publication No. WO 2007/032498)
- N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (X-a) (7.4 g).
1H-NMR (DMSO-d6) δ : 1. 45-1. 54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J = 7.8 Hz), 7.46 (2H, d, J = 8.2 Hz), 8.01 (2H, d, J = 1.8 Hz), 8.46 (1H, t, J = 1.8 Hz), 8.75 (1H, d, J = 6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s).
MS (ESI) m/z: 548 (M+H)+.
Anal.: C24H30ClN7O4S·C7H8O3S·H2O
Theoretical: C; 50.43, H; 5.46, N; 13.28, Cl; 4.80, S; 8.69.
Found: C; 50.25, H; 5.36, N; 13.32, Cl; 4.93, S; 8.79. mp (dec.): 245-248°C.

PATENT
http://www.google.com/patents/EP2589590A1?cl=en
- A compound represented by the following formula (X) [hereinafter, also referred to as compound (X)] or a pharmacologically acceptable salt thereof, or a hydrate thereof is a compound that exhibits an FXa inhibitory effect, as disclosed in Patent Literatures 1 to 3, and is useful as a preventive and/or therapeutic drug for thrombotic and/or embolic diseases:
- The pamphlet of International Publication No. WO 2007/032498discloses a process for preparing an FXa inhibitor compound (X) or a pharmacologically acceptable salt thereof, or a hydrate thereof. The process for producing compound (X) disclosed therein involves, as shown in [Scheme A] below, azidifying compound (2) to produce azide compound (3), subsequently reducing compound (3) into amino compound (1a), subsequently treating compound (1a) with anhydrous oxalic acid to obtain compound (1), which is then treated with compound (4) (ethyl[5-chloropyridin-2-yl]amino](oxo)acetate hydrochloride) in the presence of a base to produce compound (5), followed by several steps from compound (5). This pamphlet also discloses crystals of the oxalate of compound (1) as a production intermediate.
- wherein Boc represents a tert-butoxycarbonyl group.


- Patent Literature 1: International Publication No.WO 2004/058715
- Patent Literature 2: International Publication No.WO 2003/016302
- Patent Literature 3: International Publication No.WO 2003/000680
- Patent Literature 4: International Publication No.WO 2007/032498

“First market approval in Japan for LIXIANA (Edoxaban)”. Press Release. Daiichi Sankyo Europe GmbH. 2011-04-22.
- Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (September 2008). “DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles”. J. Thromb. Haemost.6 (9): 1542–9. doi:10.1111/j.1538-7836.2008.03064.x. PMID18624979.
- Raskob, G.; Cohen, A. T.; Eriksson, B. I.; Puskas, D.; Shi, M.; Bocanegra, T.; Weitz, J. I. (2010). “Oral direct factor Xa inhibition with edoxaban for thromboprophylaxis after elective total hip replacement”. Thrombosis and Haemostasis104 (3): 642–649. doi:10.1160/TH10-02-0142.PMID20589317. edit
- “Phase III Trial Finds Edoxaban Outclasses Enoxaparin in Preventing Venous Thromboembolic Events”. 8 Dec 2010.
- Weitz JI, Connolly SJ, Patel I, Salazar D, Rohatagi S, Mendell J, Kastrissios H, Jin J, Kunitada S (September 2010). “Randomised, parallel-group, multicentre, multinational phase 2 study comparing edoxaban, an oral factor Xa inhibitor, with warfarin for stroke prevention in patients with atrial fibrillation”. Thromb. Haemost.104 (3): 633–41. doi:10.1160/TH10-01-0066.
- Edoxaban versus Warfarin in Patients with Atrial Fibrillation Robert P. Giugliano, M.D., Christian T. Ruff, M.D., M.P.H., Eugene Braunwald, M.D., Sabina A. Murphy, M.P.H., Stephen D. Wiviott, M.D., Jonathan L. Halperin, M.D., Albert L. Waldo, M.D., Michael D. Ezekowitz, M.D., D.Phil., Jeffrey I. Weitz, M.D., Jindřich Špinar, M.D., Witold Ruzyllo, M.D., Mikhail Ruda, M.D., Yukihiro Koretsune, M.D., Joshua Betcher, Ph.D., Minggao Shi, Ph.D., Laura T. Grip, A.B., Shirali P. Patel, B.S., Indravadan Patel, M.D., James J. Hanyok, Pharm.D., Michele Mercuri, M.D., and Elliott M. Antman, M.D. for the ENGAGE AF-TIMI 48 InvestigatorsDOI: 10.1056/NEJMoa1310907
- “Edoxaban versus Warfarin for the Treatment of Symptomatic Venous Thromboembolism”. N. Engl. J. Med. August 2013. doi:10.1056/NEJMoa1306638. PMID23991658.
- WO 03/000657 pamphlet WO 03/000680 pamphlet WO 03/016302 pamphlet WO 04/058715 pamphlet WO 05/047296 pamphlet WO 07/032498 pamphlet WO 08/129846 pamphlet WO 08/156159 pamphlet
- J Am Chem Soc 1978, 100(16): 5199
| [1] | 王利华, 赵丽嘉, 李文利, 等. 直接抑制凝血因子Xa 的口服抗凝药物Edoxaban Tosilate Hydrate [J]. 药物评价研究, 2011, 34(6): 478-481. |
| [2] | Ohta T, Komoriya S, Yoshino T, et al. Preparation of N,N'-bis( heterocyclicacyl) cycloalkanediamine and heterocyclediamine derivatives as inhibitors of activated blood coagulation factor X (factor Xa): WO, 2003 000657 [P]. 2003-01-03. (CA 2003, 138: 73271) |
| [3] | Ohta T, Komoriya S, Yoshino T, et al. Preparation of heterocyclic moiety-containing diamine derivatives as FXa inhibitors: WO, 2003 000680 [P]. 2003-01-03. (CA 2003, 138: 89801) |
| [4] | Mochizuki A, Nagata T. Triamine derivative: WO, 2006106963 [P]. 2005-03-31. (CA 2006, 145: 419128) |
| [5] | Kawanami K, Ishikawa H, Shoji M. Process for preparation of optically active (1S,3R,4R)-3-amino-4-hydroxy-N,Ndimethylcyclohexanecarboxamide derivative salt: WO, 2012002538 [P]. 2012-01-05. (CA 2012, 156: 122056) |
| [6] | Sato K, Kubota K. Process for producing optically active carboxylic acid: WO, 2010067824 [P]. 2010-06-17. (CA 2010, 153: 36882) |
| [7] | Yoshikawa K, Yokomizo A, Naito H, et al. Design, synthesis, and SAR of cis-1,2-diaminocyclohexane derivatives as potent factor Xa inhibitors. Part I: Exploration of 5-6fused rings |
| [8] | as alternative S1 moieties [J]. Bioorg Med Chem, 2009, 17(24): 8206-8220. |
| [9] | Sato K, Kawanami K, Yagi T. Process for the preparation of optically active cyclohexane-1,2-diamine derivative from 7-oxabicyclo[4.1.0]heptane compound: WO, 2007032498 |
| [10] | 2007-03-22. (CA 2007, 146: 358502) |
| [11] | Kawanami K. Method for the preparation of optically active diamine derivative: WO, 2010104106 [P]. 2010-09-16. (CA 2010, 153: 406061) |
| [12] | Koyama T, Kondo S. Process for the preparation of diamine derivative: WO, 2010104078 [P]. 2010-09-16. (CA 2010, 153: 382938) |
| [13] | Suzuki T, Ono M. Crystal of diamine derivative and method of producing same: WO, 2011115066 [P]. 2011-09-22. (CA 2011, 155: 467954) |
| US8357808 | 9 Sep 2011 | 22 Jan 2013 | Daiichi Sankyo Company, Limited | Process for producing diamine derivative |
| US8394821 | 13 Jul 2011 | 12 Mar 2013 | Daiichi Sankyo Company, Limited | Activated blood coagulation factor inhibitor |
| US8404847 | 17 Jun 2011 | 26 Mar 2013 | Daiichi Sankyo Company, Limited | Method for producing diamine derivative |
| US8449896 | 16 Dec 2011 | 28 May 2013 | Daiichi Sankyo Company, Limited | Pharmaceutical composition having improved solubility |
| US8541443 | 19 Sep 2012 | 24 Sep 2013 | Daiichi Sankyo Company, Limited | Crystal of diamine derivative and method of producing same |
| US20130004550 * | 22 Aug 2012 | 3 Jan 2013 | Daiichi Sankyo Company, Limited | Sustained-release solid preparation for oral use |
| WO2014081047A1 | 22 Nov 2013 | 30 May 2014 | Daiichi Sankyo Company,Limited | Process for the preparation of (1s,4s,5s)-4-bromo-6-oxabicyclo[3.2.1] octan-7-one |
| Molecular Formula | C24H30ClN7O4S.C7H7HSO3 |
| Molecular Weight | 720.26 |
| CAS Registry Number | 480449-71-6 (912273-65-5) |
- N1-(5-chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide p-toluenesulfonic acid monohydrate represented by the following formula (A) (hereinafter, also referred to as compound A) :


- is known as a compound that exhibits an inhibitory effect on activated blood coagulation factor X (FXa), and is useful as a preventive and/or therapeutic drug for thrombotic diseases (Patent Literature 1 to 8).
- For example, a method comprising mixing the free form of compound A represented by the following formula (B) (hereinafter, also referred to as compound B):

- with p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate, followed by crystallization from aqueous ethanol, is known as a method for obtaining compound A (Patent Literature 1 to 8). These literature documents do not make any mention about adding p-toluenesulfonic acid or p-toluenesulfonic acid monohydrate in a stepwise manner in the step of obtaining compound A from compound B.
- Patent Literature 1: International Publication No. WO 03/000657
- Patent Literature 2: International Publication No. WO 03/000680
- Patent Literature 3: International Publication No. WO 03/016302
- Patent Literature 4: International Publication No. WO 04/058715
- Patent Literature 5: International Publication No. WO 05/047296
- Patent Literature 6: International Publication No. WO 07/032498
- Patent Literature 7: International Publication No. WO 08/129846
- Patent Literature 8: International Publication No. WO 08/156159
OTHER SALTS
Edoxaban hydrochloride
CAS Number: 480448-29-1
Molecular Formula: C24H30ClN7O4S · HCl
Molecular Weight: 584.52 g.mol-1
Edoxaban is reported to be a member of the so-called “Xaban-group” and as such to be a low molecular inhibitor of the enzyme factor Xa, participating in the blood coagulation system. Therefore, edoxaban is classified as an antithrombotic drug and its possible medical indications are reported to be treatment of thrombosis and thrombosis prophylaxis after orthopaedic operations, such as total hip replacement, as well as for stroke prevention in patients with atrial fibrillation, the prophylaxis of the acute coronary syndrome and the prophylaxis after thrombosis and pulmonary embolism.
The IUPAC name for edoxaban is N’-(5-chloropyridin-2-yl)-N-[(15,2^,4S)-4- (dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[l ,3]thiazolo[5,4-c]pyridine-2- carbonyl)amino]cyclohexyl]oxamide. The chemical structure of edoxaban is shown in the formula (1) below:
2-amino-5-methyl-4,5,6,7-tetrahydro thiazolone [5,4-c] pyridine
WO2015125710
(Reference Example 1) 2-amino-5-methyl-4,5,6,7-tetrahydro thiazolone [5,4-c] pyridine (1-n) (The method described in WO 2005/047296 Pamphlet )
[0091]
[Of
35] in 2-PrOH (1.44L) solution was heated to 50 ℃ 1-
methyl-4-piperidone (180.0g), 2-PrOH (360mL) solution of cyanamide
(67.0g), and sulfur powder (51.0 g) it was added. Pyrrolidine (13.3mL)
was added to the reaction mixture, after stirring for 2 hours at 50 ℃,
followed by stirring overnight and allowed to cool to room
temperature. The reaction mixture was cooled to 10 ℃ less in an ice
water bath and stirred for 1 hour at the same temperature. Is filtered
and the precipitated crystals were washed with 2-PrOH (540mL), the title
compound was dried under reduced pressure at 40 ℃ (209.9g, 78%) was
obtained.
[0092]
1 H-NMR (CDCl 3 ) ppm: 4.86 (Br, 2H), 3.47-3.46 (t, 2H, J = 1.9 Hz), 2.78-2.71 (M, 2H), 2.71-2.65 (M, 2H), 2.47 . (s, 3H)
MS (FAB) M / z: 170 (M + H) +
elemental analysis: C 7 H 11 N 3 as S,
theoretical value: C, 49.68; H, 6.55; N, 24.83; S, 18.95
measured value: C, 49.70; H, 6.39; N, 24.91; S, 19.00.
MS (FAB) M / z: 170 (M + H) +
elemental analysis: C 7 H 11 N 3 as S,
theoretical value: C, 49.68; H, 6.55; N, 24.83; S, 18.95
measured value: C, 49.70; H, 6.39; N, 24.91; S, 19.00.
(Example 11) N 1 - (5-Chloro-2-yl) -N 2 -
[(1S, 2R, 4S)-4-(dimethylcarbamoyl) -2 - {[(5-methyl-4,5 ,
6,7-tetrahydro [1,3] thiazolo [5,4-c] pyridin-2-yl) carbonyl] amino}
cyclohexyl] Etanjiamido (X) [Production method via Compound (1-p2)]
[0137]
In
10 mL test tube, compound (5-ms) (the compound of Reference Example 8)
(100 mg, 0.216 mmol), Compound (1-p2) (81.4 mg, 0.216 mmol), K 3 PO 4 (91.7 mg, 0.432 mmol) and DMF (1 mL) was added, and the mixture was stirred at room temperature conditions for 3 hours. H To the reaction mixture 2 O
(2 mL) was added and the resulting slurry was stirred at room
temperature overnight, the solid was filtered. The resulting solid H 2 was washed with O (1 mL), was obtained by drying under reduced pressure the title compound (110.0 mg, 92.9%) as a solid.
[0138]
1 H-NMR (500 Hz, CDCl 3 )
delta: 9.72 (s, 1H), 8.30 (dd, 1H, J = 2.5, 0.5 Hz), 8.17 (dd, 1H, J =
9.0, 0.5 Hz), 8.03 (D , 1H, J = 8.5 Hz), 7.68 (dd, 1H, J = 9.0, 2.5 Hz),
7.39 (d, 1H, J = 8.5 Hz), 4.70-4.67 (m, 1H), 4.13-4.09 (m, 1H), 3.73
(d, 1H, J = 16.0 Hz), 3.70 (d, 1H, J = 16.0 Hz), 3.06 (s, 3H), 2.96-2.93
(m, 2H), 2.95 (s, 3H), 2.89-2.79 (m, 3H), 2.52 (s, 3H), 2.14-2.06 (m,
3H), 1.96-1.90 (m, 1H), 1.84-1.78 (m, 1H), 1.69-1.62 (m, 1H ).
Edoxaban, DU-176b
SYN..........http://newdrugapprovals.org/2015/01/13/daiichi-sankyo-receives-fda-approval-for-anti-clotting-drug-savaysa-edoxaban/
.............
13 C NMR
FREE BASE
- (Reference
Example 6)
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide
(X) (production method described in the pamphlet of International
Publication No. WO 2007/032498)

- Methanesulfonic acid (66 ml) was added to a suspension of tert-butyl [(1R,2,S,5S)-2-({[(5-chloropyridin-2-yl)amino](oxo)acetyl}amino)-5-(dimethylaminocarbonyl)cyclohexyl]carbamate (5) (95.1 g) in acetonitrile (1900 ml) at room temperature, and the mixture was stirred at this temperature for 2 hours. To the reaction solution, triethylamine (155 ml), 5-methyl-4,5,6,7-tetrahydro[1,3]thiazolo[5,4-c]pyrzdine-2-carboxylic acid hydrochloride (8) (52.5 g), 1-hydroxybenzotriazole (33.0 g), and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (46.8 g) were added under ice cooling, and the mixture was stirred at room temperature for 16 hours. Triethylamine and water were added thereto, and the mixture was stirred for 1 hour under ice cooling. Then, crystals were collected by filtration to obtain the title compound (X) (103.2 g).
- 1H-NMR (CDCl3) δ : 1.60-1.98 (3H, m), 2.00-2.16 (3H, m), 2.52 (3H, s), 2.78-2.90 (3H, m), 2.92-2.98 (2H, m), 2.95 (3H, s), 3.06 (3H, s), 3.69 (1H, d, J = 15.4 Hz), 3.75 (1H, d, J = 15.4 Hz), 4.07-4.15 (1H, m), 4.66-4.72 (1H, m), 7.40 (1H, dd, J = 8.8, 0.6 Hz), 7. 68 (1H, dd, J = 8.8, 2.4 Hz), 8.03 (1H, d, J = 7.8 Hz), 8.16 (1H, dd, J = 8.8, 0.6 Hz), 8.30 (1H, dd, J = 2. 4, 0.6 Hz), 9.72 (1H, s).
- MS (ESI) m/z: 548 (M+H)+.
| Molecular Formula | C24H30ClN7O4S.C7H7HSO3 | |
| Molecular Weight | 720.26 | |
| CAS Registry Number | 480449-71-6 (912273-65-5) |
TOSYLATE
- (Reference
Example 7)
N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide
mono-p-toluenesulfonate monohydrate (X-a) (production method described
in the pamphlet of International Publication No. WO 2007/032498)

- N1-(5-Chloropyridin-2-yl)-N2-((1S,2R,4S)-4-[(dimethylamino)carbonyl]-2-{[(5-methyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridin-2-yl)carbonyl]amino}cyclohexyl)ethanediamide (X) (6.2 g) was dissolved in methylene chloride (120 ml). To the solution, a 1 mol/L solution of p-toluenesulfonic acid in ethanol (11.28 ml) was added, and the solvent was distilled off. To the residue, 15% hydrous ethanol (95 ml) was added, and the mixture was dissolved by stirring at 60°C. Then, the mixture was cooled to room temperature and stirred for 1 day. The precipitated crystals were collected by filtration, washed with ethanol, and then dried under reduced pressure at room temperature for 2 hours to obtain the title compound (X-a) (7.4 g).
- 1H-NMR (DMSO-d6) δ : 1. 45-1. 54 (1H, m), 1.66-1.78 (3H, m), 2.03-2.10 (2H, m), 2.28 (3H, s), 2.79 (3H, s), 2.91-3.02 (1H, m), 2.93 (3H, s), 2.99 (3H, s), 3.13-3.24 (2H, m), 3.46-3.82 (2H, m), 3.98-4.04 (1H, m), 4.43-4.80 (3H, m), 7.11 (2H, d, J = 7.8 Hz), 7.46 (2H, d, J = 8.2 Hz), 8.01 (2H, d, J = 1.8 Hz), 8.46 (1H, t, J = 1.8 Hz), 8.75 (1H, d, J = 6.9 Hz), 9.10-9.28 (1H, br), 10.18 (1H, br), 10.29 (1H, s).
MS (ESI) m/z: 548 (M+H)+.
Anal.: C24H30ClN7O4S·C7H8O3S·H2O
Theoretical: C; 50.43, H; 5.46, N; 13.28, Cl; 4.80, S; 8.69.
Found: C; 50.25, H; 5.36, N; 13.32, Cl; 4.93, S; 8.79. mp (dec.): 245-248°C.
1H NMR PREDICTION, TOSYLATE
CAS NO. 1229194-11-9, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate H-NMR spectral analysis
CAS NO. 1229194-11-9, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate H-NMR spectral analysis
13 CNMR PREDICTION, TOSYLATE
CAS NO. 1229194-11-9, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate C-NMR spectral analysis
![N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate NMR spectra analysis, Chemical CAS NO. 1229194-11-9 NMR spectral analysis, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate C-NMR spectrum](https://images-blogger-opensocial.googleusercontent.com/gadgets/proxy?url=http%3A%2F%2Fpic11.molbase.net%2Fnmr%2Fnmr_image%2F2015-01-11%2F001%2F717%2F1717235_13c.png&container=blogger&gadget=a&rewriteMime=image%2F*)
CAS NO. 1229194-11-9, N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide,4-methylbenzenesulfonic acid,hydrate C-NMR spectral analysis
SYN..........http://newdrugapprovals.org/2015/01/13/daiichi-sankyo-receives-fda-approval-for-anti-clotting-drug-savaysa-edoxaban/
..................
WO2015125710
(Example 11) N 1 - (5-Chloro-2-yl) -N 2 -
[(1S, 2R, 4S)-4-(dimethylcarbamoyl) -2 - {[(5-methyl-4,5 ,
6,7-tetrahydro [1,3] thiazolo [5,4-c] pyridin-2-yl) carbonyl] amino}
cyclohexyl] Etanjiamido (X) [Production method via Compound (1-p2)]
[0137]
In
10 mL test tube, compound (5-ms) (the compound of Reference Example 8)
(100 mg, 0.216 mmol), Compound (1-p2) (81.4 mg, 0.216 mmol), K 3 PO 4 (91.7
mg, 0.432 mmol) and DMF (1 mL) was added, and the mixture was stirred
at room temperature conditions for 3 hours. H To the reaction mixture 2 O
(2 mL) was added and the resulting slurry was stirred at room
temperature overnight, the solid was filtered. The resulting solid H 2 was washed with O (1 mL), was obtained by drying under reduced pressure the title compound (110.0 mg, 92.9%) as a solid.
[0138]
1 H-NMR (500 Hz, CDCl 3 )
delta: 9.72 (s, 1H), 8.30 (dd, 1H, J = 2.5, 0.5 Hz), 8.17 (dd, 1H, J =
9.0, 0.5 Hz), 8.03 (D , 1H, J = 8.5 Hz), 7.68 (dd, 1H, J = 9.0, 2.5 Hz),
7.39 (d, 1H, J = 8.5 Hz), 4.70-4.67 (m, 1H), 4.13-4.09 (m, 1H), 3.73
(d, 1H, J = 16.0 Hz), 3.70 (d, 1H, J = 16.0 Hz), 3.06 (s, 3H), 2.96-2.93
(m, 2H), 2.95 (s, 3H), 2.89-2.79 (m, 3H), 2.52 (s, 3H), 2.14-2.06 (m,
3H), 1.96-1.90 (m, 1H), 1.84-1.78 (m, 1H), 1.69-1.62 (m, 1H ).
References
- "First market approval in Japan for LIXIANA (Edoxaban)". Press Release. Daiichi Sankyo Europe GmbH. 2011-04-22.
- O'Riordan, Michael (9 January 2015). "FDA Approves Edoxaban for Stroke Prevention in AF and DVT/PE Prevention". Medscape. Retrieved 10 January 2015.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/206316lbl.pdf
- lexicomp.com
- Savaysa (edoxaban) [prescribing information]. Parsippany, NJ: Daiichi Sankyo; January 2015.
- http://www.drugs.com/cons/edoxaban.html
- Yoshiyuki, I., et al. "Biochemical and pharmalogical profile of darexaban, an oral direct Xa inhibitor." European Journal of Pharmacology (2011): 49-55
- Katsung, B., S. Masters and A. Trevor. Basic and Clinical Pharmacology 11th Edition. United States of America: McGraw-Hill, 2009
- Turpie AG (January 2008). "New oral anticoagulants in atrial fibrillation". European Heart Journal 29 (2): 155–65. doi:10.1093/eurheartj/ehm575. PMID 18096568.
 |
SAVAYSA is available for oral administration as a 60 mg, 30 mg, or 15 mg round shaped, film-coated tablet, debossed with product identification markings. Each 60 mg tablet contains 80.82 mg edoxaban tosylate monohydrate equivalent to 60 mg of edoxaban. Each 30 mg tablet contains 40.41 mg edoxaban tosylate monohydrate equivalent to 30 mg of edoxaban. Each 15 tablet contains 20.20 mg edoxaban tosylate monohydrate equivalent to 15 mg of edoxaban.
The inactive ingredients are: mannitol, pregelatinized starch, crospovidone, hydroxypropyl cellulose, magnesium stearate, talc, and carnauba wax. The color coatings contain hypromellose, titanium dioxide, talc, polyethylene glycol 8000, iron oxide yellow (60 mg tablets and 15 mg tablets), and iron oxide red (30 mg tablets and 15 mg tablets).
 | |
| Systematic (IUPAC) name | |
|---|---|
N'-(5-chloropyridin-2-yl)-N-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide
| |
| Clinical data | |
| Trade names | Lixiana, Savaysa |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 62%; Tmax 1–2 hours |
| Protein binding | 55% |
| Metabolism | Minimal hepatic |
| Biological half-life | 10–14 hours |
| Excretion | 50% renal; <50% bile |
| Identifiers | |
| CAS Registry Number | 912273-65-5 |
| ATC code | None |
| PubChem | CID: 25022378 |
| IUPHAR/BPS | 7575 |
| ChemSpider | 8456212 |
| UNII | NDU3J18APO |
| KEGG | D09710 |
| ChEBI | CHEBI:85973 |
| Chemical data | |
| Formula | C24H30ClN7O4S |
| Molecular mass | 548.056 g/mol |
5
APIXABAN
 APIXABAN
APIXABANApixaban
CAS 503612-47-3
APPROVALS
EMA————MAY 18, 2011
FDA…………………DEC28, 2012
PMDA………….. DEC25, 2012
CFDA………………JAN 22, 2013

Apixaban, sold under the tradename Eliquis, is a anticoagulant for the treatment of venous thromboembolic events. It is taken by mouth. It is a direct factor Xa inhibitor.
Apixaban was approved in Europe in 2012.[1] It was approved in the U.S. in 2014 for treatment and secondary prophylaxis of deep vein thrombosis (DVT) and pulmonary embolism (PE).[2] It is being developed in a joint venture by Pfizer and Bristol-Myers Squibb.[3][4]
Medical uses
Apixaban is indicated for the following:[5]- To lower the risk of stroke and embolism in patients with nonvalvular atrial fibrillation.
- Deep vein thrombosis (DVT) prophylaxis. DVT's may lead to pulmonary embolism (PE) in knee or hip replacement surgery patients.
- Treatment of both DVT and PE.
- To reduce the risk of recurring DVT and PE after initial therapy.
Atrial fibrillation
Apixaban is recommended by the National Institute for Health and Clinical Excellence for the prevention of stroke and systemic embolism in people with non-valvular atrial fibrillation and at least one of the following risk factors: prior stroke or transient ischemic attack, age 75 years or older, diabetes mellitus, or symptomatic heart failure.[6]Apixaban and other newer anticoagulants (dabigatran and rivaroxaban) appear equally effective as warfarin in preventing non-hemorrhagic stroke in people with atrial fibrillation and are associated with lower risk of intracranial bleeding.[7]
Mechanism of action
Apixaban is a highly selective, orally bioavailable, and reversible direct inhibitor of free and clot-bound factor Xa. Factor Xa catalyzes the conversion of prothrombin to thrombin, the final enzyme in the coagulation cascade that is responsible for fibrin clot formation.[10] Apixaban has no direct effect on platelet aggregation, but by inhibiting factor Xa, it indirectly decreases clot formation induced by thrombin.[5]FDA approval
A new drug application (NDA) for the approval of apixaban was submitted to the FDA by Bristol-Myers Squibb and Pfizer jointly after conclusion of the ARISTOTLE clinical trial in 2011.[11]Apixaban was approved for the prevention of stroke in people with atrial fibrillation on December 28, 2012.[12] On March 14, 2014, it was approved for the additional use of preventing deep vein thrombosis and pulmonary embolism in people that had recently undergone knee or hip replacement.[13] On August 21, 2014, the FDA approved apixaban for the treatment of recurring deep vein thrombosis and pulmonary embolism.[2]
During development it was known as BMS-562247-01.
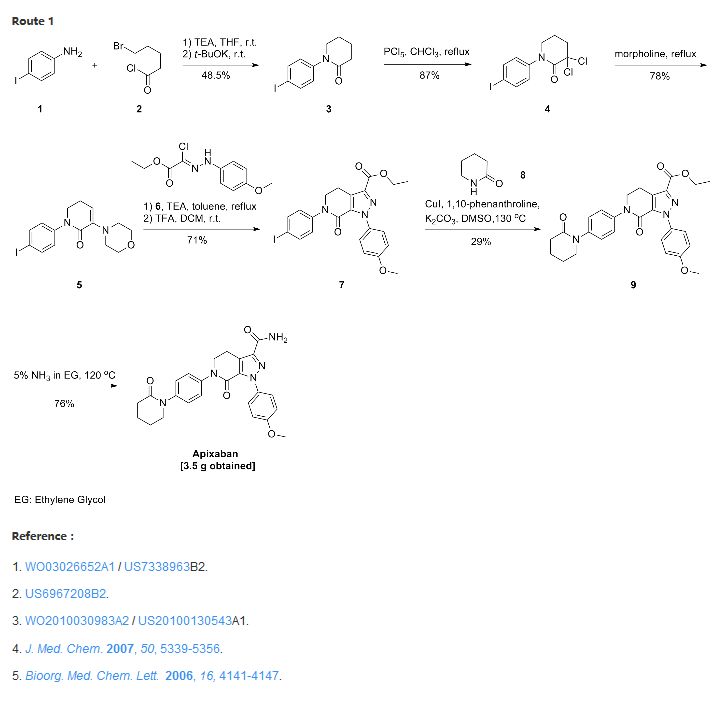






Thursday, August 21, 2014 – Bristol-Myers Squibb Company (NYSE: BMY) and Pfizer Inc. (NYSE: PFE) today announced the U.S. Food and Drug Administration (FDA) has approved a Supplemental New Drug Application (sNDA) for Eliquis for the treatment of DVT and PE, and for the reduction in the risk of recurrent DVT and PE following initial therapy. Combined, DVT and PE are known as VTE. It is estimated that every year, approximately 900,000 Americans are affected by DVT and PE.
http://www.drugs.com/newdrugs/fda-approves-eliquis-apixaban-deep-vein-thrombosis-pulmonary-embolism-4073.html?utm_source=ddc&utm_medium=email&utm_campaign=Today%27s+news+summary+-+August+21%2C+2014 –
See more at: http://worlddrugtracker.blogspot.in/2014/08/fda-approves-eliquis-apixaban-for.html

PREDICTIONS
1H NMR


13C NMR
COSY

1H NMR PREDICT


13 C NMR PREDICT


![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide H-NMR spectrum](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_vqrxkRFOvg4pABBAg2OKyh_rRQ0aJcGF8Twrs82EGNp3Fb7f6KEZO1_mqNNWAtj2Bc0aOgZMwRK-KliteWr92gZ1Tt8UuQAkEBU59k7OqgC61nO78-peF2wNauzHy_40XUwkiJLM7bXO-h0kq9U800IIgh=s0-d) CAS NO. 503612-47-3, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide H-NMR spectral analysis ![1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide NMR spectra analysis, Chemical CAS NO. 503612-47-3 NMR spectral analysis, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectrum](https://lh3.googleusercontent.com/blogger_img_proxy/AEn0k_vU3DudXZzSDc46alX16nlDRLDYiiruEkymsGWGIKQg-upmeOd3JfgU6KsHn3Xutra-NWzAVbgjcmPIq3zjRQjrcpdXIqtkqzAgIirSv6HfWZnxtecEzWJMCTm2sgSR72BJYjy2shuY_oEBJ3TTBh5iHVfdCA=s0-d) |
C-NMR spectral analysis
CAS NO. 503612-47-3, 1-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-1-yl)phenyl]-4,5-dihydropyrazolo[3,4-c]pyridine-3-carboxamide C-NMR spectral analysis http://www.google.com/patents/WO2012168364A1?cl=en
l-(4-Methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l -yl)phenyl]-4, 5,6,7- tetrahydro- lH-pyrazolo[3,4-c]pyridine-3-carboxyamide of formula (I), also known come apixaban, is a powerful inhibitor of coagulation factor Xa disclosed in US 6,967,208. Said compound is used in the prevention and treatment of thromboembolic disorders.

(I)
US 7, 153,960 discloses a process for the preparation of apixaban wherein the key step is the formation of intermediate (A) by 1 ,3 dipolar cycloaddition reaction between the compounds of formula (B) and (C) and its subsequent conversion to the compound of formula (D) by treatment with an acid. The compound of formula (D), after simple manipulations of functional groups, is converted to apixaban

B C A D
Said patent discloses the preparation of the compounds of formula (B) and (C). While the synthesis of the hydrazone of formula (B) has been known for some time, the preparation of the key intermediate of formula (C) is complex and uses reagents which are expensive and potentially hazardous, such as phosphorus pentachloride (PC15), and drastic reaction conditions.
US 7, 153,960, for example, discloses as preferred the preparation of an enamine intermediate of formula (C) wherein the amine residue NRbRc is a morpholine. The conditions used for the success of the reaction actually involve the use of morpholine as solvent at high temperatures, such as reflux temperature (about 130- 135°C).
The complexity of the known processes for the preparation of the intermediate of formula C, the expense and danger of the reagents and the drastic reaction conditions used make said processes difficult to apply and scale up industrially, especially for the purpose of preparing the intermediates of formula A and D and apixaban.
Example 6. Synthesis of compound of formula (I): l-(4- Methoxyphenyl)-6-[4-(2-oxo-piperidinyl)phenyl]-7-oxo-4,5,6,7-tetrahydro- l//-pyrazolo[3,4-c]pyridine-3-carboxyamide: Apixaban (I)

The compound of formula II, prepared as in Example 5 (17.50 g, 35.82 mmol), is suspended in 100 ml of 33% NH3 and 200 ml of MeOH in a 1L 4-necked flask equipped with coolant, thermometer and magnetic stirrer, in nitrogen atmosphere, and heated to 45°. MeOH (250 ml) is added until completely dissolved, and the solution is left under stirring for 2h. Another addition of 33% NH3 (50 ml) is performed, and the progress of the reaction is monitored by TLC (AcOEt/MeOH 9: 1) and HPLC. After 18h the solvent is evaporated under low pressure, and the solid residue obtained is suspended in 200 ml of H2O and left under stirring for 2h. The white solid is filtered through a Buchner funnel, and washed with H2O (50 ml). The product of formula (I) is stove-dried at 50°C to a constant weight (12.60 g, yield 76%). The HPLC purity of the product exceeds 99%

.
1H NMR (300 MHz, CDC13): DELTA
7.47 (2H, dd, J0=8.7 Hz, Ar-H),
7.31(2H, dd, J0=8.7 Hz, Ar-H),
7.23 (2H, dd, J0=8.7 Hz, Ar-H),
6.93 (2H, dd, J0=8.7 Hz, Ar-H),
6.83 (1H, s, N-H),
5.53 (1H, s, N-H),
4.1 1 (2H, t, J=6.6 Hz, CH2CH2N),
3.81 (3H, s, Ar-OCH3),
3.59 (2H, m, NCH2CH2CH2CH2CO)
3.37 (2H, t, J=6.6 Hz, CH2CH2N),
2.55 (2H, m, NCH2CH2CH2CH2CO),
1.93 (4H, m, NCH2CH^CH2CH2CO).
SEE
NMR
For apixaban (in CDCl3) 1 HNMR (CDCl3) δ: 7.49 (d, J = 8.80 Hz, 2H), 7.37 (d, J= 9.10 Hz, 2H), 7.26 (d, J= 8.80 Hz, 2H), 6.98 (s, 1H), 6.95 (d, J = 9.20 Hz, 2H), 6.28 (s, 1H), 4.14 (t, J= 6.60 Hz, 2H), 3.81 (s, 3H), 3.61 (m, 2H), 3.39 (t, J = 6.60 Hz, 2H), 2.63 (t, J= 6.20 Hz, 2H), 1.96 (m, 4H) ppm FT-IR spectral data of apixaban Compound IR (KBr) absorption bands (cm−1 ) Apixaban 1630 for -C=O stretch cyclic amide; 1595 for -C=O stretch amide lactum; 3023 for -C-H stretch aromatic 
Zhou , J. C. ; Oh , L. M. ; Ma , P. ; Li , H. Y. Synthesis of 4,5-dihydro-pyrazolo[3,4-c]pyrid-2-ones. WO Patent 2003/0 49681, June 19 , 2003 .
 
J. Med. Chem. 2007 , 50 , 5339 – 5356 .
1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (1)[ref ]J. Med. Chem. 2007 , 50 , 5339 – 5356 .
To the advanced intermediate 2 (2.44 g, 5.0 mmol) was added 25% ammonia water (1.5 mL, 20 mmol) in methanol (20 mL), and the mixture was heated to 65 °C for 5 h in an autoclave of 50 mL. The resulting mixture was cooled to room temperature, poured into water (30 mL), and crystalized below 0°C. The precipitate was filtrated and dried in vacuo at 50°C to afford the desired product 1 as a pale white solid. Yield: 2.09 g, 91%; mp 171–173 °C; IR (KBr, cm−1): 3448 and 3298 (N-H stretching), 2940 (C-H aliphatic), 1669 (C˭N stretching), 1614 (C˭O stretching), 1544 (aliphatic C˭C), 1513, 1463 and 1441 (aromatic C˭C), 1334, 1300 and 1254 (C-N stretching), 1146, 1111, 1090 and 1024 (C-O stretching), 835, 816, 794 and 758 (Ar-H aromatic bending); 1H NMR (500 MHz, CDCl3, ppm), δ: 7.48 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.0 Hz, 2H), 5.66 (brs, 2H), 4.12 (t, J = 5.6 Hz, 2H), 3.84 (s, 3H), 3.55–3.65 (m, 2H), 3.39 (t, J = 5.6 Hz, 2H), 2.57 (t, J = 6.2 Hz, 2H), 1.91–2.01 (m, 4H); 13C NMR (125 MHz, CDCl3, ppm), δ: 170.9, 164.4, 160.5, 158.0, 142.1, 140.6 (2C), 134.0, 133.2, 127.4 (4C), 126.9 (2C), 126.5, 114.4 (2C), 56.2, 52.3, 51.8, 33.5, 24.2, 22.1, 21.9; MS/EI m/z = 459.2 (M+).
SEE
http://www.google.com/patents/WO2014056434A1?cl=en
....................... Patent http://www.google.com/patents/WO2014108919A2?cl=en
HPLC method of Analysis:
Apixaban compound of formula- 1 of the present invention is analyzed by HPLC using the following conditions:
Apparatus: A liquid chromatographic system is to be equipped with variable wavelength UV- detector; Column: Zorbax Bonus RP, 250 x 4.6 mm, 5μιη or equivalent; Flow rate: 1.2 ml/min; wavelength: 270 nm; column temperature: 40°C; Injection volume; 5 uL; Run time: 35 minutes; Needle wash: diluent; Diluent: Acetonitrile: water (90: 10 v/v); Elution: Gradient; Mobile phase-A: Buffer; Mobile phase-B: acetonitrile:water (90:10 v/v); Buffer: Weigh accurately about 1.36 g of potassium dihydrogen ortho phosphate in 1000 10 ml of milli-Q water and adjust pH 6.0 with dil KOH solution, then filter through 0.22 μιη nylon membrane filter paper. The following impurities have been observed during the preparation of Apixaban.

methyl esterImpurity Chloro Impurity Dehydro Impurity
Scheme-I:
Apixaban

Scheme-II:

Pure Apixaban Formula-1 [Apixaban]
Example-1: Preparation of 3-chloro-l-(4-iodophenyI)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (4.08 gm) followed by lithium chloride (2.28 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (30 gm) and dimethylformamide (60 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 25.0 gm; MR: 120-130°C.
Example-2: Preparation of 3-chIoro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Lithium carbonate (2.99 gm) followed by sodium chloride (2.76 gm) were added to a mixture of 3,3-dichloro-l-(4-iodophenyl)piperidin-2-one compound of formula-5 (50 gm) and dimethylformamide (150 ml) at 30-35°C and stirred for 10 min at the same temperature. Heated the reaction mixture to 110-115°C and stirred for 6 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 1 hr at the same temperature. Filtered the precipitated solid and then dried to get the title compound.
Yield: 42.0 gm; M.R: 120-130°C.
Example-3: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (5.09 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydro pyridin-2(lH)-one compound of formula-6 (5 gm) and toluene (5 ml) at 25-30°C and stirred for 5 min at the same temperature. Heated the reaction mixture to 115-120°C and stirred for 3 hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water was added to the reaction mixture at 25-30°C and stirred for 15 hrs at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 3.8 gm.
Example-4: Preparation of l-(4-iodophenyl)-3-morpholino-5,6-dihydropyridin-2(lH)-one (Formula-7)
Morpholine (28.73 gm) was added to a mixture of 3-chloro-l-(4-iodophenyl)-5,6- dihydropyridin-2(lH)-one compound of formula-6 (50 gm) and toluene (50 ml) at 30-35°C. Heated the reaction mixture to 115-120°C and stirred for 8 hrs at 115-120°C. After completion of the reaction, cooled the reaction mixture to 25-30°C. Methyl tert-butyl ether (100 ml) followed by water were slowly added to the reaction mixture at 25-30°C. Cooled the reaction mixture to 5- 10°C and stirred for 2 hours at 5-10°C. Filtered the precipitated solid and then dried to get the title compound. Yield: 45 gm.
Example-5: Preparation of ethyl 6-(4-iodophenyl)-l-(4-methoxyphenyI)-7-oxo-4,5,6,7-tetra hydro-lH-pyrazoIo[3,4-c]pyridine-3-carboxyIate (FormuIa-13)
A mixture of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one compound of formula-6 (79.2 gm), (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula-9 (65 gm) and toluene (450 ml) was heated to 90-100°C and stirred for 5 min at the same temperature. Triethyl amine (72 gm) was slowly added to the reaction mixture at 95-100°C and stirred for 2½ hrs at the same temperature. Cooled the reaction mixture to 25-30°C. Water (110 ml) was added to the reaction mixture at 25-30°C and stirred for 8 hrs at the same temperature. Filtered the solid, washed with water and then dried to get the title compound.
Yield: 78.5 gm.
Example-6: Preparation of 5-bromo-N-(4-iodophenyl)pentanamide (Formula-3)
A mixture of 5-bromopentanoic acid (54 g), thionyl chloride (41 g), dimethylformamide (2 ml) and toluene (100 ml) was heated to 40-45°C and stirred for 2 hours at the same temperature. Distilled off the reaction mixture to remove the un-reacted thionyl chloride under reduced pressure at a temperature below 40°C. Toluene (50 ml) was added to the reaction mixture and stirred for 15 minutes. The reaction mixture was cooled to 25-30°C under nitrogen atmosphere and it slowly added to a pre-cooled mixture of 4-iodoaniline compound of formula-2 (50 g) and toluene (350 ml) at 0-5°C. Triethyl amine (29 g) was added to it at 0-5°C. The above reaction mixture containing acid chloride was slowly added to the reaction mixture containing 4- iodoaniline under nitrogen atmosphere and stirred for 2 hours at 0-5°C. Water (250 ml) was added to the reaction mixture and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and then dried to get title compound. Yield: 83 gm; MR: 135-140°C; HPLC purity: 99%.
Example-7: Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula-6)
Step-a) Preparation of l-(4-iodophenyl)piperidin-2-one (Formula-4)
Sodium tert-butoxide (18.86 g) was added to a mixture of 5-bromo-N-(4- iodophenyl)pentanamide compound of formula-3 (50 g) and toluene (250 ml) at 0-5°C and stirred for 2 hours at 0-5°C. Water (100 ml) followed by aqueous hydrochloric acid solution (50 ml) were added to the reaction mixture and stirred for 10 minutes at 5-10°C. Both the organic and aqueous layers were separated; the organic layer was washed with water. Distilled off the solvent from the organic layer under reduced pressure at a temperature below 60°C to get title compound as a solid.
Step-b) Preparation of 3,3-dichIoro-l-(4-iodophenyI)piperidin-2-one (Formula-5)
The compound obtained in step-a) was dissolved in dichloromethane (100 ml) and slowly added to a mixture of phosphorous pentachloride (95 g) and dichloromethane (150 ml) at 25- 30°C. The reaction mixture was heated to 35-40°C and stirred for 4 hours at the same temperature. Cooled the reaction mixture to 5-10°C. Chilled water (150 ml) was added to the reaction mixture and stirred for 1.5 hours at 10-15°C. Both the organic and aqueous layers were separated; the organic layer was washed with water followed by 10% aqueous sodium carbonate solution. Distilled off the solvent completely from the organic layer to get title compound as a solid.
Step-c) Preparation of 3-chloro-l-(4-iodophenyl)-5,6-dihydropyridin-2(lH)-one (Formula- 6)
To the obtained compound in step-b), dimethylformamide (100 ml), followed by lithium carbonate (2.2 g) and sodium chloride (2.0 g) were added at 25-30°C. The reaction mixture was heated to 115-120°C and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 30-35°C, water (350 ml) was added to it and stirred for 2 hours at 25-30°C. Filtered the precipitated solid and washed with water. Methanol (360 ml) was added to the obtained solid and the reaction mixture was heated to 65-70°C. Stirred the reaction mixture for 20 minutes at the same temperature. Carbon (3.0 g) was added to the reaction mixture and stirred for 20 minutes at 65-70°C. Filtered the reaction mixture through hyflow bed and washed with methanol. Distilled off the solvent from the filtrate under reduced pressure and methanol (300 ml) was added to the residue and stirred for 20 minutes at 25-30°C. Cooled the reaction mixture to -5 to 0°C and stirred for 60 minutes at the same temperature. Filtered the precipitated solid, washed with methanol and then dried to get title compound.
Yield: 25 gm; MR: 115- 120°C: HPLC purity: 98%.
Example-8: Preparation of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydro pyridin-2(lH)-one (Formula-8)
A mixture of l-(4-iodophenyl)-3-mo holino-5,6-dihydropyridin-2(lH)-one compound of formula-7 (50 g), piperidin-2-one (32.25 g) and o-xylene (75 ml) was stirred for 10 minutes at 25-30°C. Potassium carbonate (27.0 g), followed by copper iodide (7.43 g) were added to the reaction mixture. The reaction mixture was heated to 140-145°C under azeotropic distillation condition and stirred for 6 hours at the same temperature. Cooled the reaction mixture to 35- 40°C, water (175 ml) was slowly added to the reaction mixture at 35-40°C. Cooled the reaction mixture to 10-15°C and ammonia (125 ml) was added to the reaction mixture at 10-15°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with water and then dried to get title compound.
Yield: 35 gm; MR: 195-200°C; HPLC purity: 95%.
Example-9: Preparation of (Z)-ethyl 2-chloro-2-(2-(4-nlethoxyphenyl)hydrazono)acetate (FormuIa-9)
A mixture of 4-methoxyaniline compound of formula- 12 (50 g) and water (150 ml) was cooled to 5-10°C. Hydrochloric acid (100 ml), followed by a solution of sodium nitrite (30.81 g) in water (50 ml) were slowly added to the reaction mixture at 5-10°C and stirred for 2 hours at 5- 10°C to provide diazotized compound. Ethyl acetate (250 ml) was added to the reaction mixture. Ethyl 2-chloro acetoacetate (76.84 g) was slowly added to a mixture of sodium acetate (76.6 g), ethyl acetate (250 ml) and water (150 ml) at 25-30°C and the reaction mixture was stirred for 2 hours at 25-30°C. The reaction mixture was slowly added to the reaction mixture containing diazotized compound at a temperature below 10°C. The temperature of the reaction mixture was raised to 25-30°C and stirred for 16 hours at the same temperature. Both the organic and aqueous layers were separated and the organic layer was washed with 10% aqueous sodium bicarbonate solution followed by 10% aqueous sodium chloride solution. Distilled off the solvent completely from the organic layer under reduced pressure and then co-distilled with toluene. Toluene was added to the obtained compound and stirred for 15 minutes at 25-30°C. Silica-gel was added to the reaction mixture and stirred for 30 minutes at 25-30°C. Filtered the reaction mixture and the solvent from the filtrate was distilled off completely under reduced pressure. Cyclohexane (400 ml) was added to the obtained compound and the reaction mixture was stirred for 60 minutes at 25-30°C. Filtered the precipitated solid, washed with cyclohexane and then dried to get title compound. Yield: 60 gm; MR: 95-100°C; HPLC purity: 99%.
ExampIe-10: Preparation of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl) phenyl)-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate (Formula-11)
A mixture of 3-morpholino-l-(4-(2-oxopiperidin-l-yl)phenyl)-5,6-dihydropyridin-2(lH)- one compound of formula-8 (30 g), sodium carbonate (26.83 g) and acetone (150 ml) was heated to 45-50°C. (Z)-ethyl 2-chloro-2-(2-(4-methoxyphenyl)hydrazono)acetate compound of formula- 9 (32.5 g) was added to the reaction mixture at 45-50°C and stirred for 3 hours at the same temperature. Cooled the reaction mixture to 25-30°C and aqueous hydrochloric acid (50 ml) in 50 ml of water was added to it at 25-30°C. Stirred the reaction mixture for 2 hours at 25-30°C. Water was slowly added to the reaction mixture and stirred for 45 minutes at 25-30°C. Filtered the obtained solid and washed with water. The obtained solid was recrystallized from toluene (150 ml) to get the title compound. Yield: 35 gm; MR: 155-160°C; HPLC purity: 97%.
Example- 11: Preparation of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
A mixture of ethyl l-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-l-yl)phenyl)- 4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxylate compound of formula-11 (50 g), formamide (150 ml), sodium methoxide (30 ml) and isopropanol (300 ml) was heated to 65-70°C and stirred for 2 hours at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid and washed with isopropanol. Methanol (150 ml) was added to the obtained solid, the reaction mixture was heated to 65-70°C and stirred for 15 minutes at 65-70°C. Cooled the reaction mixture to 0-5°C and stirred for 30 minutes at 0-5°C. Filtered the precipitated solid, washed with methanol and then dried to get title compound. Yield: 35 g. MR: 230-235°C; HPLC purity: 98%.
The PXRD of the crystalline solid obtained from the above example is matches with the PXRD of crystalline form-M of the present invention.
Example-12: Purification of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]- 4,5,6, 7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (100 g) was dissolved in a mixture of dichloromethane (1200 ml) and methanol (200 ml) at 25-30°C. 10% aqueous sodium carbonate solution (200 ml) was added to the reaction mixture and stirred for 15 minutes at 25- 30°C. Both the organic and aqueous layers were separated, methanol (100 ml) was added to the organic layer and again 200 ml of 10% aqueous sodium carbonate solution was added to the reaction mixture. The reaction mixture was stirred for 15 minutes at 25-30°C and separated the organic and aqueous layers. To the organic layer methanol (100 ml) followed by water (200 ml) were added. Both the organic and aqueous layers were separated. The solvent from organic layer was distilled under reduced pressure at a temperature below 40°C. 3000 ml of a mixture of dichloromethane and methanol (in the ratio of 3:7) was added to the crude compound and the reaction mixture was heated to reflux temperature and stirred for 10 minutes. Carbon (10 g) was added to the reaction mixture and stirred for 15 minutes at the reflux temperature. Filtered the reaction mixture through hyflow bed, washed with a mixture of dichloromethane and methanol. The filtrate was cooled to 0-5°C and stirred for 2 hours at 0-5°C. Filtered the precipitated solid and washed with a mixture of dichloromethane and methanol. Isopropanol (1000 ml) was added to the reaction mixture. Heated the reaction mixture to 80-85°C and stirred for 15 minutes. Cooled the reaction mixture to 25-30°C and stirred for 2 hours at 35-30°C. Filtered the precipitated solid, washed with isopropanol and then dried to get title compound.
Yield: 80 gm; MR: 235-240°C.
The PXRD pattern of crystalline solid obtained from the above example is matches with PXRD of crystalline form-M of the present invention.
Example-13: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to isopropanol (400 ml) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min the same temperature. Filtered the solid, washed with isopropanol and then dried to get the title compound. Yield: 4.5 gm; Water content: 0.30% w/w. HPLC purity: 99.8%; Acid impurity: 0.02%; Amino acid impurity: Not detected; Chloro impurity: 0.01%; Methyl ester impurity: 0.05%; Ethyl ester impurity: 0.01%; Dehydro impurity: 0.07%.
Particle size distribution: D(0.1): 9.183 μπι; D(0.5): 25.991 um; D(0.9): 60.749 μιη; D[4,3]: 31.066 μπι.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-14: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yI)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (6.25 gm) was added to 50% aqueous isopropanol (60 ml) at 25-30°C. Heated the reaction mixture to 50-60°C and stirred for 4 hrs at the same temperature. Cooled the reaction mixture to 25-30°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get the title compound.
Yield: 4.1 gm; Water content: 0.35% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-15: Preparation of crystalline form-S of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0-5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and then dried to get the title compound. Yield: 24.0 gm; M.R: 235-245°C; Water content: 7.38% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-3 and figure-4 respectively.
Example-16: Preparation of crystalline form-N of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3- carboxamide(Formula-l)
A mixture of dichloromethane and ethyl acetate (625 ml, in 3:7 ratio) was added to l-(4- methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c] pyridine-3-carboxamide compound of formula- 1 (6.25 gm) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Cooled the reaction mixture to 0-5°C and stirred for 60 min at the same temperature. Filtered the solid and then dried to get title compound. Yield: 3.9 g; Water content: 5.21% w/w.
The PXRD and DSC of the obtained compound are illustrated in figure-5 and figure-6 respectively.
Example-17: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
1 -(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin- 1 -yl)phenyl]-4,5,6,7-tetrahydro- 1 H- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to isopropanol (510 ml). Heated the reaction mixture to reflux temperature and stirred for 15 Minutes at the same temperature. The reaction mixture was cooled to 0-5°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1. Yield: 23 g; Water content: 0.30%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively.
Example-18: Preparation of crystalline form-M of l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxo piperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH-pyrazolo[3,4-c]pyridine-3-carboxamide (Formula-1)
l-(4-methoxyphenyl)-7-oxo-6-[4-(2-oxopiperidin-l-yl)phenyl]-4,5,6,7-tetrahydro-lH- pyrazolo[3,4-c]pyridine-3-carboxamide compound of formula-1 (34 gm) was added to a mixture of dichloromethane and methanol (1020 ml, in 3:7 ratio) at 25-30°C. Heated the reaction mixture to reflux temperature and stirred for 15 min at the same temperature. Filtered the reaction mixture and washed with a mixture of dichloromethane and methanol. Cooled the filtrate to 0- 5°C and stirred for 60 min at the same temperature. Filtered the precipitated solid and added to aq.isopropanol (340 ml). Heated the reaction mixture to 50-60°C and stirred for 15 minutes at the same temperature. The reaction mixture was cooled to 25-35°C and stirred for 60 minutes at the same temperature. Filtered the solid and then dried to get crystalline form-M of compound of formula-1.
Yield: 23 g; Water content: 0.35%w/w.
The PXRD and DSC of the obtained compound are illustrated in figure- 1 and figure-2 respectively
/.....................
|
References
- "ELIQUIS® (apixaban) Approved In Europe For Preventing Venous Thromboembolism After Elective Hip Or Knee Replacement" (Press release). Pfizer. April 20, 2012. Retrieved 2012-05-29.
- http://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_eliquis_apixaban_for_the_treatment_of_deep_vein_thrombosis_dvt_and_pulmonary_embolism_pe_and_for_the_reduction_in_the_risk_of_recurrent_dvt_and_pe_following_initial_therapy
- "Bristol-Myers Squibb News Release 26 April 2007". Archived from the original on 11 September 2007. Retrieved 2007-09-15.
- Nainggolan, Lisa. "Apixaban better than European enoxaparin regimen for preventing VTE". Retrieved 2011-04-01.
- "Eliquis (apixaban) [prescribing information]" (PDF). Princeton, NJ: Bristol-Myers Squibb. March 2014. Retrieved 2014-10-29.
- "www.nice.org.uk".
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). "Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.". Thrombosis 2013: 640723. PMID 24455237.
- "ELIQUIS® (apixaban) tablets Factor Xa Inhibitor" (PDF). FDA. August 2014. Retrieved 2014-11-02.
- Enriquez A, Lip GY, Baranchuk A (2015). "Anticoagulation reversal in the era of the non-vitamin K oral anticoagulants". Europace. doi:10.1093/europace/euv030. PMID 25816811.
- Frost C, Wang J, Nepal S; et al. (February 2013). "Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects". Br J Clin Pharmacol 75 (2): 476–87. doi:10.1111/j.1365-2125.2012.04369.x. PMID 22759198.
- Granger, M.D. et. al., Christopher (September 15, 2011). "Apixaban versus Warfarin in Patients with Atrial Fibrillation". New England Journal of Medicine (365): 981–992. doi:10.1056/NEJMoa1107039. Retrieved 17 September 2015.
- FDA approves Eliquis to reduce the risk of stroke, blood clots in patients with non-valvular atrial fibrillation
6
7
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
 COCK WILL TEACH YOU
COCK WILL TEACH YOU
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE


 amcrasto@gmail.com
amcrasto@gmail.com
/////////////////////
No comments:
Post a Comment