
1 Montelukast
2 Pranlukast
3 Zafirlukast
4 Ibudilast
5
will be updated

 MONTELUKAST
MONTELUKAST
MK-0476 (Montelukast, L-706631)
US 8,007,830, US 5,565,473*PED, MERCK
Pat exp...Aug 3, 2012
| NPP | Mar 26, 2015 |
NDA 020829, 20/2/98, SINGULAIR, tablet oral, merck

Montelukast
CAS : 158966-92-8
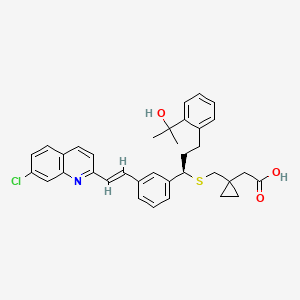
CAS Name: 1-[[[(1R)-1-[3-[(1E)-2-(7-Chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl]cyclopropaneacetic acid
Molecular Formula: C35H36ClNO3S
Molecular Weight: 586.18
Percent Composition: C 71.71%, H 6.19%, Cl 6.05%, N 2.39%, O 8.19%, S 5.47%
Derivative Type: Monosodium salt
Sodium 1-(((1(R)-(3 -(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methylethyl)phenyl)propyl)thio)methyl)cyclopropane-acetate
1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)-thio)methyl)cyclopropylacetate sodium salt
CAS Registry Number: 151767-02-1
Manufacturers' Codes: MK-476
Trademarks: Singulair (Merck & Co.)
Molecular Formula: C35H35ClNNaO3S
Molecular Weight: 608.17
Percent Composition: C 69.12%, H 5.80%, Cl 5.83%, N 2.30%, Na 3.78%, O 7.89%, S 5.27%
Properties: Hygroscopic, white to off-white powder. Freely sol in ethanol, methanol, water. Practically insol in acetonitrile.
Therap-Cat: Antiasthmatic.
Antiasthmatic (Nonbronchodilator); Leukotriene Antagonist.
Montelukast is a leukotriene receptor antagonist (LTRA) used for the maintenance treatment of asthma and to relieve symptoms of seasonal allergies. It is usually administered orally. Montelukast blocks the action of leukotriene D4 on the cysteinyl leukotriene receptor CysLT1 in the lungs and bronchial tubes by binding to it. This reduces the bronchoconstriction otherwise caused by the leukotriene, and results in less inflammation. Because of its method of operation, it is not useful for the treatment of acute asthma attacks. Again because of its very specific locus of operation, it does not interact with other allergy medications such as theophylline. Montelukast is marketed in United States and many other countries by Merck & Co. with the brand name Singulair®. It is available as oral tablets, chewable tablets, and oral granules. In India and other countries, it is also marketed under the brand name Montair®, produced by Indian company Cipla.
MONTELUKAST (Singulair® Oral Granules) helps to reduce asthma symptoms (coughing, wheezing, shortness of breath, or chest tightness) and control your asthma. It does not provide instant relief and cannot be used to treat a sudden asthma attack. It works only when used on a regular basis to help reduce inflammation and prevent asthma attacks. This drug is also helpful in improving seasonal allergies, like hay fever.
Montelukast is effective in adults and children
Amongst the US approvals, tentative FDA approvals have been identified for generic Montelukast sodium, awarded to Endo, Glenmark, Mylan, Roxane, Sandoz, Teva and Torrent. The large number of generic authorisations awaiting launch in the UK is indicative of the likely competition the Singulair product will face across Europe upon SPC expiry
EP Pat. No. 480,717 discloses Montelukast sodium along with other related compounds and the methods for their preparation. The reported method of synthesis proceeds through corresponding methyl ester namely, Methyl 2-[(3S)-[3-[(2E)-(7-chloroquinolin - 2yl) ethenyl] phenyl] – 3 – hydroxypropyl] benzoate and involves coupling methyl 1- (mercaptomethyi) cyclopropaneacetate with a mesylate generated in-situ.
The methyl ester of Montelukast is hydrolyzed to free acids and the latter converted directly to Montelukast Sodium salt (Scheme -1). The process is not particularly suitable for large – scale production because it requires tedious chromatographic purification of the methyl ester intermediate and / or the final product and the product yield is low.
Scheme -1

U.S. Pat. No. 5,614632 disclosed a process for the preparation of crystalline Montelukast sodium, which comprises of the following steps (Scheme – 2):
■ Reaction of methyl 2-[3(S)-[3-[2-(7-chloroquinolin -2-yl) ethenyl] phenyl] -3- hydroxypropyl benzoate (I) with Grignard reagent, methyl magnesium chloride in presence of cerium chloride to give Diol (II) ■ Reaction of Diol (II) with methane sulfonyl chloride to afford 2-[2-[3 (s)-[3- (2-(7-chloro quinolin-2yl) ethenyl] phenyl]- 3 – methane sulfonyloxy propyl] phenyl] -2-propanol (III)
■ Condensation of 2-[2-[3(s)-[3-(2-(7-chloro quinolin - 2-yl) ethenyl] phenyl] -
3 – methane sulfonyloxypropyl] phenyl] – 2- propanol (III) with dilithium anion of 1-mercaptomethyl) cyclopropaneacetic acid, which has been generated by the reaction of l-(mercaptomethyl)cyclopropaneacetic acid (IV)with n-Butyl lithium
■ Isolation of the condensed product, Montelukast as solid Montelukast dicyclohexylamine salt
■ Purification and conversion of Montelukast dicyclohexylamine salt into Montelukast sodium
■ Crystallization of Montelukast sodium from a mixture of toluene and acetonitrile
The process disclosed in U.S Pat. No. 5,614,632 further involved the reaction of Diol (II) with methane sulfonyl chloride involves the reaction temperature of about – 25°C and the storage condition of the intermediate, 2-[2-[3(s)-[3-(2-(7-chloro quinolin - 2-yl) ethenyl] pheny] -3 -methane sulfonyloxy propyl] phenyl] -2-propanol (III) at temperature below – 150C for having the stability. The process further involves the reaction, formation of dilithium anion of l-(mercaptomethyl) cyclopropaneacetic acid which requires the usage of n-Butyl lithium, a highly flammable and hazardous reagent and the reaction is at temperature below -5°C further requires anhydrous conditions. Scheme – 2




Montelukast (trade names Singulair, Montelo-10, and Monteflo and Lukotas in India) is a leukotriene receptor antagonist(LTRA) used for the maintenance treatment of asthma and to relieve symptoms of seasonal allergies.[1][2] It is usually administered orally once a day. Montelukast is a CysLT1 antagonist; it blocks the action of leukotriene D4 (and secondary ligands LTC4 and LTE4) on the cysteinyl leukotriene receptor CysLT1 in the lungs and bronchial tubes by binding to it. This reduces the bronchoconstriction otherwise caused by the leukotriene and results in less inflammation.
Because of its method of operation, it is not useful for the treatment of acute asthma attacks. Again because of its very specificmechanism of action, it does not interact with other asthma medications such as theophylline.
Another leukotriene receptor antagonist is zafirlukast (Accolate), taken twice daily. Zileuton (Zyflo), an asthma drug taken four times per day, blocks leukotriene synthesis by inhibiting 5-lipoxygenase, an enzyme of the eicosanoid synthesis pathway.
Singulair was covered by U.S. Patent No. 5,565,473[9] which expired on August 3, 2012.[10] The same day, the FDA approved several generic versions of montelukast.[11]
On May 28, 2009, the United States Patent and Trademark Office announced their decision to launch a reexamination of the patent covering Singulair. The decision to reexamine was driven by the discovery of references that were not included in the original patent application process. The references were submitted through Article One Partners, an online research community focused on finding literature relating to existing patents. The references included a scientific article produced by a Merck employee around the key ingredient of Singulair, and a previously filed patent in the same technology area.[12]
On December 17, 2009, the U.S. Patent and Trademark Office determined that the patent in question was valid based on the initial reexamination and new information provided.[13]
Montelukast is currently available in film coated tablet and orodispersible tablet formulations for once-daily administration, and also available as an oral granule formulation which is specifically designed for administration to paediatric patients.
Patent family US17493193A claims crystalline Montelukast sodium and processes for its preparation . Patents within this family are not considered to be a constraint to generic competition because the protected technology may possibly be circumvented by the synthesis and use of different molecular forms and/or salts. Patent family US33954901P relates to the specific marketed oral granule formulation of Montelukast.
The chemical name of montelukast sodium is: Sodium 1-[[[(1R)-1-[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl]cyclopropaneacetic acid and its structure is represented as follows:

- Montelukast is apparently a selective, orally active leukotriene receptor antagonist that inhibits the cysteinyl leukotriene CysLT1 receptor.
- The chemical name for montelukast sodium is [R-(E)]-1-[[[1-[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl] cyclopropaneacetic acid, monosodium salt. Montelukast sodium salt is understood to be represented by the following structural formula:

- U.S. patent No. 5,565,473 (“’473 patent”) is listed in the FDA’s Orange Book for montelukast sodium. The ’473 patent recites a broad class of leulcotriene antagonists as “anti-asthmatic, anti-allergic, anti-inflammatory, and cycloprotective agents” represented by a generic chemical formula. ’473 patent, col. 2,1. 3 to col. 4,1. 4. Montelukast is among the many compounds represented by that formula. The ’473 patent also refers to pharmaceutical compositions of the class of leukotriene antagonists of that formula with pharmaceutically acceptable carriers. Id. at col. 10,11. 42-46.
- Montelukast sodium is currently marketed by Merck in the form of film coated tablets and chewable tablets under the trade name Singular®. The film coated tablets reportedly contain montelukast sodium and the following inactive ingredients: microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, hydroxypropylcellulose, magnesium stearate, titanium dioxide, red ferric oxide, yellow ferric oxide, and carnauba wax. The chewable tablets reportedly containmontelukast sodium and the following inactive ingredients: mannitol, microcrystalline cellulose, hydroxypropylcellulose, red ferric oxide, croscarmellose sodium, cherry flavor, aspartame, and magnesium stearate. Physicians’ Desk Reference, 59th ed. (2005), p. 2141.
- However, there is a need in the art to improve the stability of compositions of montelukast and particularly those of the sodium salt.
Montelukast sodium is a leukotriene antagonist and inhibits the leukotriene biosynthesis. It is a white to off-white powder that is freely soluble in methanol, ethanol, and water and practically insoluble in acetonitrile.
A montelukast sodium salt is a substance which exhibits efficacy of Singulair (available from Korean MSD) generally used for the treatment of asthma as well as for the symptoms associated with allergic rhinitis, which is pharmaceutically known as a leukotriene receptor antagonist. Leukotrienes produced in vivo by metabolic action of arachidonic acid include LTB4, LTC4, LTD4 and LTE4. Of these, LTC4, LTD4 and LTE4 are cysteinyl leukotrienes (CysLTs), which are clinically essential in that they exhibit pharmaceutical effects such as contraction of airway muscles and smooth muscles and promotion of secretion of bronchial mucus.
Montelukast sodium salt is a white and off-white powder which has physical and chemical properties that it is well soluble in ethanol, methanol and water and is practically insoluble in acetonitrile.
A conventionally known method for preparing a montelukast sodium salt is disclosed in EP Patent No. 480,717. However, the method in accordance with the EP Patent requires processes for introducing and then removing a tetrahydropyranyl (THP) protecting group and purification by chromatography, thus being disadvantageously unsuitable for mass-production. In addition, the method disadvantageously requires investment in high-cost equipment, for example, to obtain amorphous final compounds by lyophilization.
Meanwhile, U.S. Pat. No. 5,614,632 discloses an improved method for preparing a montelukast sodium salt by directly reacting a methanesulfonyl compound (2) with 1-(lithium mercaptomethyl)cyclopropaneacetic acid lithium salt, without using the tetrahydropyranyl protecting group used in EP Patent No. 480,717, purifying in the form of a dicyclohexylamine salt by adding dicyclohexylamine to the reaction solution, and converting the salt into a montelukast sodium salt (1).
However, the method in accordance with the US patent should use n-butyl lithium as a base in the process of preparing the 1-(lithium mercaptomethyl)cyclopropaneacetic acid lithium salt and thus requires an improved process due to drawbacks that n-butyl lithium is dangerous upon handling and is an expensive reagent.
PCT International Patent Laid-open No. WO 2005/105751 discloses a method for preparing a montelukast sodium salt, comprising coupling methyl 1-(mercaptomethyl)cyclopropane acetate (3) used in step 10 shown in Example 146 of EP Patent 480,717 with a methanesulfonyl compound (2) in the presence of a solvent/cosolvent/base, performing hydrolysis, recrystallizing the resultingmontelukast acid (4) in the presence of a variety of solvents to obtain highly puremontelukast acid (4), and converting the same into a montelukast sodium salt (1).
In addition, WO 2005/105751 claims that, in the coupling reaction, one is selected from tetrahydrofurane and dimethylcarbonate as a solvent, a highly polar solvent is selected from dimethylformamide, dimethylacetamide and N-methylpyrrolidone as a cosolvent, and one is selected from sodium hydroxide, lithium hydroxide, sodium hydride, sodium methoxide, potassium tert-butoxide, lithium diisopropylamine and quaternary ammonium salts, as a base.
However, WO 2005/105751 discloses that, since the coupling reaction requires use of a mixed solvent and the mixed solvent is different from the solvent used for hydrolysis, a process for removing the cosolvent through distillation under reduced pressure or extraction is further required prior to hydrolysis.
Further, in accordance with the method of WO 2005/105751, recrystallization is performed in the presence of a variety of solvents in order to obtain a highly puremontelukast acid (4) and the resulting recrystallization yield is varied in a range of 30 to 80%, depending on the solvent. In the case where desired purity is not obtained, recrystallization is repeated until montelukast acid (4) with a desired purity can be obtained. Disadvantageously, the method causes deterioration in overall yield.
European Patent No. 480,717 discloses montelukast sodium and its preparation starting with the hydrolysis of its ester derivative to the crude sodium salt, acidification of the crude to montelukast acid, and purification of the crude acid by column chromatography to give montelukast acid as an oil. The resulting crude oil in ethanol was converted to montelukast sodiumby the treatment with an equal molar aqueous sodium hydroxide solution. After removal of the ethanol, the montelukastsodium was dissolved in water and then freeze dried. The montelukast sodium thus obtained is of a hydrated amorphous form as depicted in FIG. 2.
The reported syntheses of montelukast sodium, as pointed out by the inventor in EP 737,186, are not suitable for large-scale production, and the product yields are low. Furthermore, the final products, as the sodium salts, were obtained as amorphous solid which are often not ideal for pharmaceutical formulation. Therefore, they discloses an efficient synthesis of montelukastsodium by the use of 2-(2-(3-(S)-(3-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-methanesulfonyloxypropyl)phenyl)-2-propanol to couple with the dilithium salt of 1-(mercaptomethyl)cyclopropaneacetic acid. The montelukast acid thus obtained is converted to the corresponding dicyclohexylamine salt and recrystallized from a mixture of toluene and acetonitrile to obtain crystallinemontelukast sodium. This process provides improved overall product yield, ease of scale-up, and the product sodium salt in crystalline form.
According to the process described in EP 737,186, the chemical as well as optical purities of montelukast sodium depends very much on the reaction conditions for the mesylation of the quinolinyl diol with methanesulfonyl chloride. For instance, the reaction temperature determinates the chemical purity of the resulting coupling product montelukast lithium, due to the fact that an increase in the reaction temperature resulted in decreased selectivity of mesylation toward the secondary alcohol. Mesylation of the tertiary alcohol occurred at higher temperature will produce, especially under acidic condition, the undesired elimination product, the styrene derivative. This styrene impurity is difficult to remove by the purification procedure using DCHA salt formation; while excess base, butyl lithium in this case, present in the reaction mixture causes the formation of a cyclization by-product, which will eventually reduce the product yield.
PCT WO 2005/105751 discloses an alternative process for preparing montelukast sodium by the coupling of the same mesylate as disclosed in ’186 patent with 1-(mercaptomethyl)cyclopropane alkyl ester in the presence of a base. In this patent, the base butyl lithium, a dangerous and expensive reagent, is replaced with other milder organic or inorganic base. However, the problem concerning the formation of the styrene impurity is still not resolved.
CA 2649189 A1
Process for the manufacture of 1-[[[(1R)-1-[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid, sodium salt [montelukast sodium (I)] consisting of: i. Converting methyl 1-(mercaptomethyl)-cyclopropaneacetate to a metal salt (X) using a metal hydroxide, ii. Subjecting the metal salt (X) to monometallation to provide a dimetallide (XI). iii. Converting a diol of formula (II) to a mesylate of formula (III) and reacting (III) in situ with (XI) affordin the metal salt of 1-[[[(1R)-1-[3-[(1E)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid. iv. Reacting the metal salt in-situ with a base and purifying to afford an amine salt (XII). v. Treating (XII) with a sodium base and precipitating out montelukast sodium (I).

more info
European Patent No. 480,717 disclose the montelukast and its preparation method first be hydrolyzed to the crude ester derivatives sodium, then this crude product was acidified to montelukast acid (montelukastacid), Finally, column chromatography purification of this crude acid into oily montelukast acid. This oilMontelukast acid in ethanol, by equimolar amounts of sodium hydroxide solution and converted to montelukast sodium.
The ethanol was removed aftermontelukast sodium dissolved in water, followed by freeze-drying. Finally obtainedmontelukast shown in Figure 2 is amorphous hydrated.
The invention, in European Patent No. 737,186 points out, thismontelukast synthesis method is not suitable for mass production, and the low yield.
Moreover, the resulting amorphous solid salt, are generally not used in pharmaceutical formulations.
Therefore, they disclose the synthesis of an effective method of montelukast sodium, which uses 2 – (2 – (3 – (S) – (3 – (7 – chloro-2 – quinolinyl) ethenyl) phenyl) -3 – methylsulfonyl) phenyl) -2 – propanol and 1 – (methylthio alcohol) cyclopropane coupling the lithium salt of acetic acid, the resulting Montelukast acid is converted into a corresponding bicyclic hexyl amine salt, and from a mixture of toluene and acetonitrile recrystallization to prepare crystalline montelukast. This method greatly improves the productivity, ease of mass production, and the product is crystalline sodium salt.
According to European Patent No. 737,186 described method for preparingmontelukast chemical purity and optical purity depends largely quinoline diol with methanesulfonyl chloride in the reaction between the mesylated condition.
For example, the reaction temperature resulted in an increase of the secondary alcohols methanesulfonyl selective reduction, the reaction temperature determines the coupling product (montelukast lithium) chemical purity. Occurs at a higher temperature mesylation tertiary alcohols, in particular under acidic conditions, will produce impurities, such as styrene derivatives.
This impurity is difficult styrene generated by using the DCHA salt (DCHA salt formation) in the purification process to remove; present in the reaction mixture and excess base, butyl lithium cyclized by-products resulting in the formation will eventually reduce the yield of the product.
W02005/105751 disclose another preparation method of montelukast sodium, which is the methanesulfonic acid (European Patent No. 737,186 is the same) in an alkaline state where 1_ (methyl mercaptan yl) cyclopropyl alkyl ester and coupling thereof. In this patent, the dangerous and expensive alkaline-butyl lithium reagent, is replaced by other more moderate organic or inorganic base. However, the formation of styrene impurity problem is still not resolved
LitReferences: Selective cysteinyl leukotriene type 1 receptor antagonist. Prepn: M. L. Belley et al., EP 480717; eidem, US 5565473 (1992, 1996 both to Merck Frosst); M. Labelle et al., Bioorg. Med. Chem. Lett. 5, 283 (1995).
Pharmacological profile: T. R. Jones et al., Can. J. Physiol. Pharmacol. 73, 191 (1995).
LC determn in human plasma: R. D. Amin et al., J. Pharm. Biomed. Anal. 13, 155 (1995).
Review of pharmacology and clinical efficacy in asthma: A. Markham, D. Faulds, Drugs 56, 251-256 (1998).
Clinical trial in pediatric asthma: B. Knorr et al., J. Am. Med. Assoc. 279, 1181 (1998); with loratadine, q.v., in allergic rhinitis: E. O. Meltzer et al., J. Allergy Clin. Immunol. 105, 917 (2000).
Comparison with cetirizine, q.v., in urticaria: M. L. Pacor et al., Clin. Exp. Allergy 31, 1607 (2001).
Review of pharmacology and clinical experience: Z. Diamant, A. P. Sampson, J. Drug Eval. Respir. Med. 1, 53-88 (2002).
- Lipkowitz, Myron A. and Navarra, Tova (2001) The Encyclopedia of Allergies (2nd ed.) Facts on File, New York, p. 178, ISBN 0-8160-4404-X
- “Asthma / Allergy “. Mascothealth.com. Retrieved 9 April 2011.
- http://www.merckfrosst.ca/mfcl/en/corporate/research/accomplishments/singulair.html
- “Montelukast Sodium”. The American Society of Health-System Pharmacists. Retrieved 3 April 2011.
- FDA Investigates Merck Drug-Suicide Link
- Updated Information on Leukotriene Inhibitors: Montelukast (marketed as Singulair), Zafirlukast (marketed as Accolate), and Zileuton (marketed as Zyflo and Zyflo CR). Food and Drug Administration. Published June 12, 2009. Accessed June 13, 2009.
- Rubenstein, Sarah (April 28, 2008). “FDA Sneezes at Claritin-Singulair Combo Pill”. The Wall Street Journal.
- Schering-Plough press release – Schering-Plough/MERCK Pharmaceuticals Receives Not-Approvable Letter from FDA for Loratadine/Montelukast
- 5,565,473
- Singular patent details
- “FDA approves first generic versions of Singulair to treat asthma, allergies”. 03 August 2012. Retrieved 15 August 2012.
- “U.S. Reexamines Merck’s Singulair Patent”. Thompson Reuters. May 28, 2009.
- “Merck Says U.S. Agency Upholds Singulair Patent”. Thompson Reuters. December 17, 2009.
updated info
GENERAL METHOD1

………………………………..
READ ABOUT S ISOMER

…………………….
PAPER
Improved Process for the Preparation of Montelukast: Development of an Efficient Synthesis, Identification of Critical Impurities and Degradants
Zentiva k.s., Department of Chemical Synthesis, U kabelovny 130, Prague 102 01, Czech Republic
Org. Process Res. Dev., 2010, 14 (2), pp 425–431
DOI: 10.1021/op900311z
Publication Date (Web): February 11, 2010

1H NMR (DMSO-d6) δ (ppm) 0.23−0.47 (m, 4H, 2 × CH2 cyclopropyl), 1.08 (d, 6H, 2 × CH3isopropyl), 1.44 (s, 6H, 2 × CH3), 2.10−2.30 (m, 4H, 2 × CH2), 2.51 (m, 1H, CH), 2.52 and 2.63 (m, 2H, CH2), 2.77 a 3.07 (2 × m, 2H, CH2), 3.06 (m, 1H, CH isopropyl), 4.01 (t, 1H, CH), 5.70 (bb, 4H, NH3+, OH), 7.03−8.41 (m, 15H, CH═CH, and CH−arom.).
HPLC
HPLC (isocratic mode) chromatograms were measured with the EliteLachrom device made by the Hitachi Company. Stationary phase: RP-18e was used for the analyses; column temperature was 20 °C. Mobile phase: Acetonitrile (80%) and a 0.1 M aqueous solution of ammonium formate adjusted to pH 3.6 with formic acid (20%) were used. The flow rate of the mobile phase was 1.5 mL/min. Detection at the wavelength of 234 nm was used. Methanol was used as the solvent for preparation of samples; 10−20 μL of the solution was used for the injection. The isocratic HPLC method was used for checking the compositions of the reaction mixtures.
HPLC (gradient mode) chromatograms were measured with the Alliance HPLC device with PDA detector. Stationary phase: STAR RP-8e, 250 mm × 4 mm, 5 μm was used for the analyses; column temperature was 15 °C. Mobile phase: Acetonitrile (A) and 0.01 M aqueous solution of KH2PO4 adjusted to pH 2.2 with phosphoric acid (B) were used. Gradient mode with the flow rate of mobile phase 0.8 mL/min was used. Composition on the start was 60% of A and 40% of B, then changed to 15% of A and 85% of B over 20 min; this composition was held for 5 min, then changed to 60% of A and 40% of B over 5 min, and this composition was held to the end (overall time 35 min.). Detection at the wavelength of 234 nm was used. Methanol was used as the solvent for the preparation of the samples; 10−20 μL of the solution was used for the injection. The gradient HPLC method was used for checking the quality of the target substance including its salts with amines and of isolated standards of impurities.
HPLC (determination of (S)-enantiomer by HPLC) chromatograms were measured with the Alliance HPLC device with PDA detector. Stationary phase: Chiralpak IA (5 μm), size 0.25 m, internal diameter 4.6 mm (manufactured by Daicel) was used for the analyses, column temperature 10 °C. Mobile phase: hexane/ethanol/1,4-dioxan/trifluoroacetic acid (77:3:20:0,1 v/v/v) was used. The flow rate of the mobile phase was 1.0 mL/min. Detection at the wavelength of 285 nm was used. Methanol was used as the solvent for preparation of samples; 10 μL of the solution was used for the injection. The isocratic elution was used for checking the optical purity of target montelukast. Typical retention times: montelukast: 9.3 min, (S)-montelukast: 12.9 min.

KEY REFERENCES
(a) Ray, U. K.;Boju, S.; Pathuri, S. R.; Meenakshisunderam, S. (Aurobindo Pharma Limited, India). PCT Patent Application WO/2008/001213, 2008.
(b) Wang, Y.; Wang, Y.; Brand, M.; Kaspi, J. (Chemagis Ltd., Israel). PCT Patent Application WO/2007/088545, 2007.
(c) Turchetta, S.;Tuozzi, A.; Ullucci, E.; de Ferra, L. (Chemi S.P.A.; Italy). European Patent Application EA 1,693,368, 2008.
(d) Srinivas, P. L.; Rao, D. R.; Kankan, R. N.; Relekar, J. P. (Cipla Limited, India). PCT Patent Application WO/2006/064269, 2006.
(e) Reguri, B. R.; Bollikonda, S.;Bulusu, V. V. N. C. S.; Kasturi, R. K.; Aavula, S. K. (Dr. Reddy’s Laboratory, India). U.S. Patent Application U.S.2005/0107612, 2005.
(f) Coppi, L.; Bartra Sanmarti, M.; Gasanz Guillen, Y.; Monsalvatje Llagostera, M.; Talavera Escasany, P. (Esteve Quimica, S.A., Spain). PCT Patent Application WO/2007/051828, 2007.
(g) Hung, J. T.; Wei, C. P. (Formosa Laboratories, Inc., Taiwan). U.S. Patent Application U.S.2008/0097104, 2008.
(h) McGarrity, J.; Bappert, E.; Belser, E. (Lonza A.G., Switzerland). PCT Patent Application WO/2008/131932, 2008.
(i) Suri, S.; Sarin, G. S.; Mahendru, M. (Morepen Laboratories Limited, India). PCT Patent Application WO/2006/021974, 2006.
(j) Avdagic, A.; Mohar, B.;Sterk, D.; Stephan, M. (Pliva-Istrazivanje Razvoj D.O.O., Croatia). PCT Patent Application WO/2006/000856, 2006.
(k) Overman, A.; Gieling, R. G.; Zhu, J.; Thijs, L. (Synthon B.V., Holland). PCT Patent Application WO/2005/105479, 2005.
(l) Shapiro, E.; Yahomoli, R.;Niddam-Hildesheim, V.; Sterimbaum, G.; Chen, K. (Teva Pharmaceuticals Industries Ltd., Israel). PCT Patent Application WO/2005/105751, 2005.
(m) Achmatowicz, O.; Wisniewski,K.; Ramza, J.; Szelejewski, W.; Szechner, B. (Zaklady Farmaceutyczne Polpharma, S.A., Poland). PCT Patent Application WO/2006/043846, 2006.
.....................

EXAMPLE 8 Sodium 1-(((1(R)-(3 -(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methylethyl)phenyl)propyl)thio)methyl)cyclopropane-acetateToluene (1000 mL) and water ((950 mL) were placed in a 12 liter extractor equipped with an overhead stirrer, a thermocouple, a nitrogen inlet and an addition funnel. With good mixing of the solvents, solid dicyclohexylamine salt of Example 7 (64.3 g, 82.16 mmol) was added via a powder funnel and toluene (260 mL) was used to rinse in the remaining solid. To the well stirred suspension, acetic acid (2 M, 62 mL, 124 mmol) was added at room temperature. After approximately 10 minutes stirring was stopped. Two clear phases (yellow organic layer and colorless aqueous layer) resulted, and the aqueous waste layer was drained off. Water (950 mL) was charged to the extractor and the layers were mixed thoroughly for approx. 10 minutes.
The agitation was stopped and the aqueous waste layer was drained off.To the organic layer (1270 mL) containing the free acid a titrated solution of sodium hydroxide in 1 % aqueous ethanol (aqueous without ethanol (0.486 M, 169 mL, 82.13 mmol) was added in a steady stream over 10 minutes at room temperature under a nitrogen atmosphere. After 10 minutes age, the clear solution of the desired sodium salt was filtered through a pad of solkafloc using toluene (100 ml) for transfer and cake wash.
The clear filtrate was transferred under nitrogen to a 3 liter, 3-necked flask equipped with an overhead stirrer, a thermocouple, a nitrogen inlet and a distillation head. The solution was concentrated under vacuum to about 400 ml (ca. 40 mm Hg, ≤40°C). The distillation head was replaced with a reflux condenser and an addition funnel. The concentrate was maintained at 40 ± 2°C and acetonitrile (400 mL) was added over 20 minutes. The clear solution was seeded with 0.5 g of the crystalline sodium salt, and the resulting mixture was maintained at 40 ± 2°C for 1.5 hours, by which time a good seed bed was established.Acetonitrile (400 ml) was slowly added over 20 minutes, maintaining the batch temperature at 40 ± 2°C. The white suspension was stirred at 40 ± 2°C for 1 hour and acetonitrile (400 mL) was slowly added over 20 minutes. The slurry was aged at 40 ± 2°C for 12 hours.
A sample of the suspension was examined by cross-polarized micro-scopy to confirm crystallinity of the solid. The suspension was cooled to room temperature and aged at room temperature for 1 hour. The crystalline sodium salt was suction filtered through a sintered funnel under nitrogen. The cake was washed with acetonitrile (400 ml). The crystalline sodium salt cake was broken up in a nitrogen glove bag and dried under vacuum with nitrogen bleed at 40-45°C. The product (49 g, 80.59 mmol, 98% yield) was packaged in a well sealed brown bottle under nitrogen. The reaction mixture and the isolated product were protected from light at all times.
- HPLC assay of the sodium salt: >99.5 A%. Chiral purity: 99.8% ee. 1H NMR (CD3OD) δ 8.23 (d, 1H), 7.95 (d, 1H), 7.83 (d, 1H), 7.82 (d, 1H), 7.75 (d, H), 7.70 (bs, 1H), 7.54 (dt, 1H), 7.46 (dd, 1H), 7.42-7.35 (m, 3H), 7.37 (d, 1H), 7.14-7.00 (m, 3H), 4.86 (s, active H), 4.03 (dd, 1H), 3.09 (m, 1H), 2.82 (m, 1H), 2.66 (d, 1H), 2.52 (d, 1H), 2.40 (d, 1H), 2.30 (d, 1H), 2.24-2.14 (m, 2H), 1.51 (two s, 6H), 0.52-0.32 (m, 4H). DSC melting endotherm with a peak temperature of 133°C and an associated heat of 25 J/g.
- X-ray powder diffraction pattern: as shown in FIGURE 3.
………………
Paper
J. Liang*, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. N. Cherat, G. G. Pai
Codexis, Inc., Redwood City, USA and Arch PharmaLabs Limited, Mumbai, India
Development of a Biocatalytic Process as an Alternative to the (-)-DIP-Cl-Mediated Asymmetric Reduction of a Key Intermediate of Montelukast
Org. Process Res. Dev. 2010, 14: 193-198
Codexis, Inc., Redwood City, USA and Arch PharmaLabs Limited, Mumbai, India
Development of a Biocatalytic Process as an Alternative to the (-)-DIP-Cl-Mediated Asymmetric Reduction of a Key Intermediate of Montelukast
Org. Process Res. Dev. 2010, 14: 193-198

Montelukast sodium (Singulair®) is a leukotriene receptor antagonist prescribed for the treatment of asthma and allergies. Workers at Codexis used directed evolution and high-throughput screening to engineer a robust and efficient ketoreductase enzyme (CDX-026) that accomplished the asymmetric reduction of ketone A, which is essentially water insoluble, at a loading of 100 g/L in the presence of ca. 70% organic solvents at 45 ˚C. The (S)-alcohol B was obtained in >95% yield in >99.9% ee and in >98.5% purity on a >500 mol scale.
The enzymatic reduction entails the reversible transfer of a hydride from isopropanol to the ketone A with concomitant formation of acetone. The reaction is driven to completion by the fortuitous crystallization of the monohydrate B. The four-step conversion of B into montelukast sodium is described in the Merck process patent (M. Bhupathy, D. R. Sidler, J. M. McNamara, R. P. Volante, J. J. Bergan US 6320052, 2001). This biocatalytic reduction is superior to the reduction of A with (-)-DIPCl previously used in the manufacture of montelukast
Impurities
Montelukast sodium (I) is an active ingredient of products used for the treatment of respiration diseases, mainly asthma and nasal allergy. Montelukast sodium, chemically the sodium salt of [R-(E)]-l-[[[l-[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(l-hydroxy-l- methylethyl)phenyl]propyl]thio]-methyl]cyclopropane acetic acid is described by the chemical formula (I).
 (I)
(I)
The first solution of chemical synthesis of montelukast (I) was described in the patent no. EP 0480717 Bl and subsequently in specialized literature as well (M.Labele, Bioorg.Med.Chem.Lett. 5 (3), 283-288 (1995)). More possibilities of chemical synthesis of montelukast (I) are described in the following patents: EP 0480717 Bl, EP 0737186 Bl, US 2005/0234241 Al, WO 2005/105751 Al, US 2005/0107612 Al, WO 2005/105749 A2, WO 2005/105750 Al, US 2007/208178 Al.
 (H) R alkyl
(H) R alkyl
 R-ι> R2 alkyl or hydrogen
R-ι> R2 alkyl or hydrogen
For the process of isolation and purification of crude montelukast salts of montelukast with some amines (II) or montelukast acid (III) in the solid state have been used so far. Among montelukast salts with amines salts with dicyclohexylamine (EP 0737186 Bl, WO 04108679A1), tert-butylamine (US 2005/0107612 Al, WO 06043846A1), ethylphenylamine (US 2005/0107612 Al), isopropylamine (WO 2007/005965 Al), di-n-propylamine (WO 2007/005965 Al) and with cycloalkylamines (C5-C9, US 2007/213365 Al) have been described. Solid forms of montelukast acid, both crystalline and amorphous, have been described in a number of patent applications: WO 2005/040123, WO 2005/073194 A2, WO 2005/074893 Al, WO 2005/074893 Al, WO 2004/108679 Al, WO 2005/074935 Al. The most common method used in practice consists in purifying crude montelukast (I) via its salts with secondary amines, mainly with dicyclohexylamine (EP 0737186 Bl).
The sodium salt of montelukast, its preparation and various forms, amorphous or crystalline, , are described in a number of patents or patent applications, e.g. amorphous montelukast sodium is dealt with by EP 0737186 Bl, WO 03/066598 Al, WO 2004/108679 Al, WO 2005/074893 Al, WO 2006/054317A1 a WO 2007/005965. Crystalline polymorphs of montelukast sodium are described by WO 2004/091618 Al and WO 2005/075427 A2.
Processes of isolation and purification of montelukast are of crucial economic significance as they make it possible to obtain a substance that can be used for pharmaceutical purposes. These processes are used to remove impurities that result from the chemical instability of montelukast as well as the instability of the raw materials used for its chemical synthesis or non-selectivity of chemical reactions, or they may be represented by residues of the raw materials used, especially solvents. There is a general rule that chemical purity of the active pharmaceutical ingredient (API) produced in the industrial scale is one of the critical parameters for its commercialization. The American Food and Drug Administration (FDA) as well as European medicament control offices require, according to the Q7A ICH (International Conference on Harmonization) instruction, that API is freed from impurities to the maximum possible extent. The reason is achieving maximum safety of using the drug in the clinical practice. National inspection and control offices usually require that the content of an individual impurity in an API should not exceed the limit of 0.1%. All the substances (generally referred to as impurities) contained in an API over the limit of 0.1% should be isolated and characterized in accordance with the ICH recommendations. It is also recommended to isolate and characterize degradation products that are generated during the storage or usability period of API (ICH Guideline, 2006). In order to obtain information about the stability of a substance and to describe degradation products so-called "stress tests" are performed. Within these tests the API is subjected to a series of critical conditions the selection of which depends on the structure of the tested API. Usually, the influence of an increased temperature, air humidity, light, oxygen and stability in a wide pH range is assessed.
In the montelukast molecule there are a number of functional groups that impair the chemical stability of this substance. Montelukast is known to be prone to several types of degradation; it is mainly the case of three kinds of chemical transformation: (a) Oxidation of the mercapto group to the sulphoxide according to equation (1),
 (b) Isomerisation at the location of the double bond from geometry (E) to (Z), or trans to cis by the effect of light according to equation (2),
(b) Isomerisation at the location of the double bond from geometry (E) to (Z), or trans to cis by the effect of light according to equation (2),
 (c) Dehydration at the location of tert. alcohol, producing the corresponding olefin according to equation (3).
(c) Dehydration at the location of tert. alcohol, producing the corresponding olefin according to equation (3).
 Literature (E.D.Nelson, J.Pharm.Sci. 95, 1527-1539 (2006), C.Dufresne, J.Org.Chem. 1996, 61(24), 8518-8525, WO 2007005965A1) describes increased sensitivity of montelukast (or rather the mercapto group, which montelukast contains) to oxygen, see equation (I)). As the main product of oxidation of montelukast (I) (E)-montelukast-sulfoxide, chemically the sodium salt of [R-(E)]]-l-[[[l-[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(l-hydroxy- l-methylethyl)-phenyl]propyl]sulfmyl]methyl]cyclopropane acetic acid, described with chemical formula (IV), is mentioned. Contamination of the product with this impurity is undesirable. For this reason the processes leading to the target substance are carried out with the exclusion of oxygen, i.e. under the protective atmosphere of an inert gas (e.g. nitrogen according to EP 0737186 Bl). (E)-Montelukast-sulfoxide (IV) has also been described as a product of the oxidative metabolism of montelukast (Balani S. K. et al: Drug Metabolism and Disposition (1997) 25 (11), 1282-87, Dufrense C: J.Org.Chem. (1996) 61(24), 8518-25).
Literature (E.D.Nelson, J.Pharm.Sci. 95, 1527-1539 (2006), C.Dufresne, J.Org.Chem. 1996, 61(24), 8518-8525, WO 2007005965A1) describes increased sensitivity of montelukast (or rather the mercapto group, which montelukast contains) to oxygen, see equation (I)). As the main product of oxidation of montelukast (I) (E)-montelukast-sulfoxide, chemically the sodium salt of [R-(E)]]-l-[[[l-[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-3-[2-(l-hydroxy- l-methylethyl)-phenyl]propyl]sulfmyl]methyl]cyclopropane acetic acid, described with chemical formula (IV), is mentioned. Contamination of the product with this impurity is undesirable. For this reason the processes leading to the target substance are carried out with the exclusion of oxygen, i.e. under the protective atmosphere of an inert gas (e.g. nitrogen according to EP 0737186 Bl). (E)-Montelukast-sulfoxide (IV) has also been described as a product of the oxidative metabolism of montelukast (Balani S. K. et al: Drug Metabolism and Disposition (1997) 25 (11), 1282-87, Dufrense C: J.Org.Chem. (1996) 61(24), 8518-25).
Exposure of montelukast to light causes its isomerization while a montelukast derivative with geometry (Z) is generated in the location of the double bond (Smith Glen A. et al: Pharm.Res. 2004, 21(9), 1539-44). The impurity resulting from photo-instability is (Z)-montelukast, chemically the sodium salt of l-[[[(lR)-l-[3-[(lZ)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]- 3-[2-(l-hydroxy-l-methylethyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid, which is described by chemical formula (V), see equation (2).
Another degradation impurity described in literature (WO 2007005965A1) is montelukast dehydrated, chemically the sodium salt of l-[[[(lR)-l-[3-[(lE)-2-(7-chloro-2- quinolinyl)ethenyl]-phenyl]-3-[2-(l-methylethenyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid, described by chemical formula (VI), see equation (3).


The first solution of chemical synthesis of montelukast (I) was described in the patent no. EP 0480717 Bl and subsequently in specialized literature as well (M.Labele, Bioorg.Med.Chem.Lett. 5 (3), 283-288 (1995)). More possibilities of chemical synthesis of montelukast (I) are described in the following patents: EP 0480717 Bl, EP 0737186 Bl, US 2005/0234241 Al, WO 2005/105751 Al, US 2005/0107612 Al, WO 2005/105749 A2, WO 2005/105750 Al, US 2007/208178 Al.


For the process of isolation and purification of crude montelukast salts of montelukast with some amines (II) or montelukast acid (III) in the solid state have been used so far. Among montelukast salts with amines salts with dicyclohexylamine (EP 0737186 Bl, WO 04108679A1), tert-butylamine (US 2005/0107612 Al, WO 06043846A1), ethylphenylamine (US 2005/0107612 Al), isopropylamine (WO 2007/005965 Al), di-n-propylamine (WO 2007/005965 Al) and with cycloalkylamines (C5-C9, US 2007/213365 Al) have been described. Solid forms of montelukast acid, both crystalline and amorphous, have been described in a number of patent applications: WO 2005/040123, WO 2005/073194 A2, WO 2005/074893 Al, WO 2005/074893 Al, WO 2004/108679 Al, WO 2005/074935 Al. The most common method used in practice consists in purifying crude montelukast (I) via its salts with secondary amines, mainly with dicyclohexylamine (EP 0737186 Bl).
The sodium salt of montelukast, its preparation and various forms, amorphous or crystalline, , are described in a number of patents or patent applications, e.g. amorphous montelukast sodium is dealt with by EP 0737186 Bl, WO 03/066598 Al, WO 2004/108679 Al, WO 2005/074893 Al, WO 2006/054317A1 a WO 2007/005965. Crystalline polymorphs of montelukast sodium are described by WO 2004/091618 Al and WO 2005/075427 A2.
Processes of isolation and purification of montelukast are of crucial economic significance as they make it possible to obtain a substance that can be used for pharmaceutical purposes. These processes are used to remove impurities that result from the chemical instability of montelukast as well as the instability of the raw materials used for its chemical synthesis or non-selectivity of chemical reactions, or they may be represented by residues of the raw materials used, especially solvents. There is a general rule that chemical purity of the active pharmaceutical ingredient (API) produced in the industrial scale is one of the critical parameters for its commercialization. The American Food and Drug Administration (FDA) as well as European medicament control offices require, according to the Q7A ICH (International Conference on Harmonization) instruction, that API is freed from impurities to the maximum possible extent. The reason is achieving maximum safety of using the drug in the clinical practice. National inspection and control offices usually require that the content of an individual impurity in an API should not exceed the limit of 0.1%. All the substances (generally referred to as impurities) contained in an API over the limit of 0.1% should be isolated and characterized in accordance with the ICH recommendations. It is also recommended to isolate and characterize degradation products that are generated during the storage or usability period of API (ICH Guideline, 2006). In order to obtain information about the stability of a substance and to describe degradation products so-called "stress tests" are performed. Within these tests the API is subjected to a series of critical conditions the selection of which depends on the structure of the tested API. Usually, the influence of an increased temperature, air humidity, light, oxygen and stability in a wide pH range is assessed.
In the montelukast molecule there are a number of functional groups that impair the chemical stability of this substance. Montelukast is known to be prone to several types of degradation; it is mainly the case of three kinds of chemical transformation: (a) Oxidation of the mercapto group to the sulphoxide according to equation (1),



Exposure of montelukast to light causes its isomerization while a montelukast derivative with geometry (Z) is generated in the location of the double bond (Smith Glen A. et al: Pharm.Res. 2004, 21(9), 1539-44). The impurity resulting from photo-instability is (Z)-montelukast, chemically the sodium salt of l-[[[(lR)-l-[3-[(lZ)-2-(7-chloro-2-quinolinyl)ethenyl]phenyl]- 3-[2-(l-hydroxy-l-methylethyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid, which is described by chemical formula (V), see equation (2).
Another degradation impurity described in literature (WO 2007005965A1) is montelukast dehydrated, chemically the sodium salt of l-[[[(lR)-l-[3-[(lE)-2-(7-chloro-2- quinolinyl)ethenyl]-phenyl]-3-[2-(l-methylethenyl)phenyl]propyl]thio]methyl]cyclopropane acetic acid, described by chemical formula (VI), see equation (3).
............................................
Recently, montelukast or its pharmaceutically acceptable salt is known to function as an antagonist and also as a biosynthesis inhibitor against leukotrienes. The sodium salt of montelukast is commercially available from Merck under the trademark of Singulair® for treating asthma.
EP 480,717 discloses a method of preparing said montelukast sodium salt: As shown in Reaction Scheme 1, methyl 1-(mercaptomethyl)cyclopropylacetate of formula (B) is coupled with the compound of formula (A) to produce the compound of formula (C) as an intermediate, and the compound of formula (C) is then hydrolyzed to obtain the free acid form thereof, followed by treating the free acid with NaOH. However, this method gives a low yield or the manufacturing cost is high.
 THP: tetrahydropyranyl
THP: tetrahydropyranyl
PPTS: Pyridinium p-toluenesulfonateIn order to solve the above-mentioned problems, EP 737,186 suggests a method as shown in Reaction Scheme 2. This method uses a methanesulfonyl compound of formula (A′) having an unprotected hydroxyl group instead of the THP-protected compound of formula (A). Further, this method uses 1-(mercaptomethyl)cyclopropylacetate dilithium salt of formula (B′) instead of methyl 1-(mercaptoethyl)cyclopropylacetate of formula (B), thereby making the subsequent deprotection step unnecessary. Subsequently, dicyclohexylamine is added to the compound of formula (C″) to produce the compound of formula (D), which is converted to the desired sodium salt.

 However, the methanesulfonyl compound of formula (A′) used in the above process as a starting material is very unstable, which makes the whole process very complicated. Namely, the reaction to produce the compound of formula (A′) must be performed at a low temperature of about −30° C. and the product is required to be kept at about −15° C. The compound of formula (A′) thus produced is unstable toward moisture and air, and therefore, the reaction thereof has to be conducted quickly under carefully controlled conditions. Also, the synthesis of the compound of formula (B′) requires the use of n-butyllithium which is very explosive and unstable toward moisture and air. Thus, the method described in Reaction Scheme is not suitable for large-scale production.
However, the methanesulfonyl compound of formula (A′) used in the above process as a starting material is very unstable, which makes the whole process very complicated. Namely, the reaction to produce the compound of formula (A′) must be performed at a low temperature of about −30° C. and the product is required to be kept at about −15° C. The compound of formula (A′) thus produced is unstable toward moisture and air, and therefore, the reaction thereof has to be conducted quickly under carefully controlled conditions. Also, the synthesis of the compound of formula (B′) requires the use of n-butyllithium which is very explosive and unstable toward moisture and air. Thus, the method described in Reaction Scheme is not suitable for large-scale production.
EP 480,717 discloses a method of preparing said montelukast sodium salt: As shown in Reaction Scheme 1, methyl 1-(mercaptomethyl)cyclopropylacetate of formula (B) is coupled with the compound of formula (A) to produce the compound of formula (C) as an intermediate, and the compound of formula (C) is then hydrolyzed to obtain the free acid form thereof, followed by treating the free acid with NaOH. However, this method gives a low yield or the manufacturing cost is high.

PPTS: Pyridinium p-toluenesulfonateIn order to solve the above-mentioned problems, EP 737,186 suggests a method as shown in Reaction Scheme 2. This method uses a methanesulfonyl compound of formula (A′) having an unprotected hydroxyl group instead of the THP-protected compound of formula (A). Further, this method uses 1-(mercaptomethyl)cyclopropylacetate dilithium salt of formula (B′) instead of methyl 1-(mercaptoethyl)cyclopropylacetate of formula (B), thereby making the subsequent deprotection step unnecessary. Subsequently, dicyclohexylamine is added to the compound of formula (C″) to produce the compound of formula (D), which is converted to the desired sodium salt.


Example 1Preparation of 2-(2-(3-(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)-phenyl)-3-diphenylphosphate oxypropyl)phenyl)-2-propanol20 g of 2-(2-(3-(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-hydroxypropyl)phenyl)-2-propanol was dissolved in 240 ml of a mixture of methylene chloride and toluene (2:1), and 7.31 ml (1.2 eq.) of triethylamine was slowly added thereto. To the resulting mixture, 13.6 ml of diphenylchlorophosphate and 1.06 g of 4-dimethylaminopyridine were sequentially added dropwise. After about 1 hr, the completion of the reaction was confirmed by thin layer chromatography (TLC). The reaction mixture was treated with 100 ml of methylene chloride and 200 ml of distilled water. With shaking, the organic layer was separated and dried over sodium sulfate, followed by removing the solvent under reduced pressure. The residue thus obtained was dissolved in 60 ml of a mixture of ethyl acetate and n-hexane (1:3), and the product was recrystallized therefrom. The crystallized product was filtered, washed with 40 ml of distilled water and dried to obtain 29.5 g (97.8%) of the title compound as a yellow solid.m.p.: 127° C.1H-NMR (300 MHz, CDCl3): δ 8.4 (1H, d), 7.94 (1H, d), 7.75 (3H, m), 6.97-7.35 (20H, m), 5.70-5.72 (1H, m), 3.02-3.09 (2H, m), 2.29-2.34 (2H, m), 1.65 (3H, s), 1.59 (3H, s).
Example 2Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropylacetic acid12.7 g of 1-(mercaptomethyl)cyclopropylacetic acid dissolved in 90 ml of dimethylformamide was slowly added to a solution of 6.26 g of 60% sodium hydride dissolved in 90 ml of dimethylformamide at 0 to 5° C. To the resulting mixture, 30 g of 2-(2-(3-(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-diphenylphosphate oxypropyl)phenyl)-2-propanol obtained in Example 1 dissolved in 120 ml of dimethylformamide was slowly added dropwise. After the temperature was slowly increased to room temperature, the reaction was run for 18 to 20 hrs. Then, the reaction mixture was neutralized with a saturated ammonium chloride aqueous solution, and treated with ethyl acetate and distilled water. With shaking, the organic layer was separated and dried over sodium sulfate, followed by removing the solvent under reduced pressure. The residue thus obtained was dissolved in 270 ml of cyclohexane, and the product was recrystallized therefrom. The crystallized product was filtered, washed and dried to obtain 22.2 g (87.1%) of the title compound as a yellow solid.
1H-NMR (300 MHz, CD3OD): δ 8.27 (1H, d), 7.98 (1H, s), 7.78 (2H, d), 7.73 (2H, d), 7.38-7.56 (6H, m), 7.07-7.14 (3H, m), 4.84 (1H, t), 3.30-3.33 (1H, m), 2.84-2.87 (1H, m), 2.52 (2H, s), 2.41 (2H, s), 2.18-2.23 (2H, m), 1.55 (6H, s), 0.37-0.52 (4H, m).
Example 3Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)-thio)methyl)cyclopropylacetate sodium saltStep 1: Preparation of methyl 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropylacetate
2.1 g of methyl 1-(acetylthiomethyl)cyclopropylacetate dissolved in 35 ml of dimethylformamide was slowly added to a solution of 0.71 g of 60% sodium hydride dissolved in 35 ml of dimethylformamide at a temperature ranging from 0 to 5° C. To the resulting mixture, 7.73 g of 2-(2-(3-(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-diphenylphosphate oxypropyl)phenyl)-2-propanol obtained in Example 1 dissolved in 35 ml of dimethylformamide was slowly added dropwise at a temperature ranging from 0 to 5° C. After about 1 hr, the reaction mixture was treated with ethyl acetate and distilled water. With shaking, the organic layer was separated and dried over sodium sulfate, followed by removing the solvent under reduced pressure to obtain 5.68 g (84.5%) of the title compound as a yellow liquid.
1H-NMR (300 MHz, CDCl3): δ 8.12 (2H, d), 7.66-7.74 (4H, m), 7.37-7.48 (6H, m), 7.12-7.20 (3H, m), 3.96 (1H, t), 3.14-3.16 (1H, m), 2.88 (1H, m), 2.53 (2H, s), 2.43 (2H, s), 1.62 (6H, d), 0.41-0.54 (4H, m).
Step 2: Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropyl acetic acid
12 g of methyl 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)cyclopropylacetate obtained in step 1 was dissolved in a mixture of 60 ml of tetrahydrofuran and 30 ml of methyl alcohol. After adjusting the temperature to 10 to 15° C., 24 g of 10% NaOH solution was slowly added to the resulting mixture. Then, the temperature was slowly increased to room temperature (24 to 27° C.), and the reaction mixture was stirred for 20 hrs. After reaction was completed, the organic layer was separated and dried, followed by removing the solvent under reduced pressure. The residue thus obtained was mixed with water layer again, and 120 ml of toluene was added thereto. Subsequently, the pH of the reaction product was adjusted to 4 by adding 300 ml of acetic acid. The organic layer was separated again and dried over sodium sulfate, followed by removing the solvent under reduced pressure. The residue thus obtained was dissolved in 96 ml of a mixture of isopropanol and distilled water (2:1), and the product was recrystallized therefrom. The crystallized product was filtered to obtain 9.82 g (83%) of the title compound as a yellow solid.
Montelukast acid
1H-NMR (300 MHz, CD3OD): δ 8.27 (1H, d), 7.98 (1H, s), 7.78 (2H, d), 7.73 (2H, d), 7.38-7.56 (6H, m), 7.07-7.14 (3H, m), 4.84 (1H, t), 3.30-3.33 (1H, m), 2.84-2.87 (1H, m), 2.52 (2H, s), 2.41 (2H, s), 2.18-2.23 (2H, m), 1.55 (6H, s), 0.37-0.52 (4H, m).
m.p.: 154° C., purity>99%
Step 3: Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)-methyl)cyclopropylacetate sodium salt
5 g of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)cyclopropylacetic acid obtained in step 2 was mixed with 10 ml of toluene, followed by removing the solvent under reduced pressure to remove the solvent. To the residue thus obtained, 14.5 ml of toluene and 13 ml of 0.5N NaOH/MeOH solution were sequentially added. The resulting mixture was stirred for 30 min, followed by removing the solvent under reduced pressure. The residue was dissolved in 10 ml of toluene and 50 ml of n-hexane, and the product was recrystallized therefrom. The crystallized product was filtered to obtain 5.1 g (98%) of the title compound as a pale yellow solid.
Montelukast sodium
1H-NMR (300 MHz, CD3OD): δ 8.29 (1H, d), 7.99 (1H, s), 7.83-7.91 (3H, m), 7.72 (1H, s), 7.49-7.52 (2H, m), 7.38-7.44 (4H, m), 7.10-7.15 (3H, m), 4.04 (1H, t), 3.08 (1H, m), 2.82 (1H, m), 2.66 (1H, d), 2.52 (1H, d), 2.43 (1H, d), 2.29 (1H, d), 2.16-2.24 (2H, m), 1.52 (6H, s), 0.33-0.52 (4H, m)

Example 2Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropylacetic acid12.7 g of 1-(mercaptomethyl)cyclopropylacetic acid dissolved in 90 ml of dimethylformamide was slowly added to a solution of 6.26 g of 60% sodium hydride dissolved in 90 ml of dimethylformamide at 0 to 5° C. To the resulting mixture, 30 g of 2-(2-(3-(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-diphenylphosphate oxypropyl)phenyl)-2-propanol obtained in Example 1 dissolved in 120 ml of dimethylformamide was slowly added dropwise. After the temperature was slowly increased to room temperature, the reaction was run for 18 to 20 hrs. Then, the reaction mixture was neutralized with a saturated ammonium chloride aqueous solution, and treated with ethyl acetate and distilled water. With shaking, the organic layer was separated and dried over sodium sulfate, followed by removing the solvent under reduced pressure. The residue thus obtained was dissolved in 270 ml of cyclohexane, and the product was recrystallized therefrom. The crystallized product was filtered, washed and dried to obtain 22.2 g (87.1%) of the title compound as a yellow solid.
1H-NMR (300 MHz, CD3OD): δ 8.27 (1H, d), 7.98 (1H, s), 7.78 (2H, d), 7.73 (2H, d), 7.38-7.56 (6H, m), 7.07-7.14 (3H, m), 4.84 (1H, t), 3.30-3.33 (1H, m), 2.84-2.87 (1H, m), 2.52 (2H, s), 2.41 (2H, s), 2.18-2.23 (2H, m), 1.55 (6H, s), 0.37-0.52 (4H, m).
Example 3Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)-thio)methyl)cyclopropylacetate sodium saltStep 1: Preparation of methyl 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropylacetate
2.1 g of methyl 1-(acetylthiomethyl)cyclopropylacetate dissolved in 35 ml of dimethylformamide was slowly added to a solution of 0.71 g of 60% sodium hydride dissolved in 35 ml of dimethylformamide at a temperature ranging from 0 to 5° C. To the resulting mixture, 7.73 g of 2-(2-(3-(S)-(3-(2-(7-chloro-2-quinolinyl)ethenyl)phenyl)-3-diphenylphosphate oxypropyl)phenyl)-2-propanol obtained in Example 1 dissolved in 35 ml of dimethylformamide was slowly added dropwise at a temperature ranging from 0 to 5° C. After about 1 hr, the reaction mixture was treated with ethyl acetate and distilled water. With shaking, the organic layer was separated and dried over sodium sulfate, followed by removing the solvent under reduced pressure to obtain 5.68 g (84.5%) of the title compound as a yellow liquid.
1H-NMR (300 MHz, CDCl3): δ 8.12 (2H, d), 7.66-7.74 (4H, m), 7.37-7.48 (6H, m), 7.12-7.20 (3H, m), 3.96 (1H, t), 3.14-3.16 (1H, m), 2.88 (1H, m), 2.53 (2H, s), 2.43 (2H, s), 1.62 (6H, d), 0.41-0.54 (4H, m).
Step 2: Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)-ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)-cyclopropyl acetic acid
12 g of methyl 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)cyclopropylacetate obtained in step 1 was dissolved in a mixture of 60 ml of tetrahydrofuran and 30 ml of methyl alcohol. After adjusting the temperature to 10 to 15° C., 24 g of 10% NaOH solution was slowly added to the resulting mixture. Then, the temperature was slowly increased to room temperature (24 to 27° C.), and the reaction mixture was stirred for 20 hrs. After reaction was completed, the organic layer was separated and dried, followed by removing the solvent under reduced pressure. The residue thus obtained was mixed with water layer again, and 120 ml of toluene was added thereto. Subsequently, the pH of the reaction product was adjusted to 4 by adding 300 ml of acetic acid. The organic layer was separated again and dried over sodium sulfate, followed by removing the solvent under reduced pressure. The residue thus obtained was dissolved in 96 ml of a mixture of isopropanol and distilled water (2:1), and the product was recrystallized therefrom. The crystallized product was filtered to obtain 9.82 g (83%) of the title compound as a yellow solid.
Montelukast acid
1H-NMR (300 MHz, CD3OD): δ 8.27 (1H, d), 7.98 (1H, s), 7.78 (2H, d), 7.73 (2H, d), 7.38-7.56 (6H, m), 7.07-7.14 (3H, m), 4.84 (1H, t), 3.30-3.33 (1H, m), 2.84-2.87 (1H, m), 2.52 (2H, s), 2.41 (2H, s), 2.18-2.23 (2H, m), 1.55 (6H, s), 0.37-0.52 (4H, m).
m.p.: 154° C., purity>99%
Step 3: Preparation of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)-methyl)cyclopropylacetate sodium salt
5 g of 1-(((1-(R)-(3-(2-(7-chloro-2-quinolidyl)ethenyl)phenyl)-3-(2-(1-hydroxy-1-methyl-ethyl)phenyl)propyl)thio)methyl)cyclopropylacetic acid obtained in step 2 was mixed with 10 ml of toluene, followed by removing the solvent under reduced pressure to remove the solvent. To the residue thus obtained, 14.5 ml of toluene and 13 ml of 0.5N NaOH/MeOH solution were sequentially added. The resulting mixture was stirred for 30 min, followed by removing the solvent under reduced pressure. The residue was dissolved in 10 ml of toluene and 50 ml of n-hexane, and the product was recrystallized therefrom. The crystallized product was filtered to obtain 5.1 g (98%) of the title compound as a pale yellow solid.
Montelukast sodium
1H-NMR (300 MHz, CD3OD): δ 8.29 (1H, d), 7.99 (1H, s), 7.83-7.91 (3H, m), 7.72 (1H, s), 7.49-7.52 (2H, m), 7.38-7.44 (4H, m), 7.10-7.15 (3H, m), 4.04 (1H, t), 3.08 (1H, m), 2.82 (1H, m), 2.66 (1H, d), 2.52 (1H, d), 2.43 (1H, d), 2.29 (1H, d), 2.16-2.24 (2H, m), 1.52 (6H, s), 0.33-0.52 (4H, m)
PATENTS
| WO1995018107A1 | Dec 22, 1994 | Jul 6, 1995 | James J Bergan | Process for the preparation of leukotriene antagonists |
| WO2004026838A1 * | Sep 11, 2003 | Apr 1, 2004 | Michiaki Adachi | Method for producing a 3,5-dihydroxy-6-heptenoate |
| WO2009111998A2 * | Mar 11, 2009 | Sep 17, 2009 | Zentiva, K.S. | Specific impurities of montelukast |
| EP0480717A1 | Oct 10, 1991 | Apr 15, 1992 | Merck Frosst Canada Inc. | Unsaturated hydroxyalkylquinoline acids as leukotriene antagonists |
| EP0480717B1 | Oct 10, 1991 | Apr 15, 1998 | Merck Frosst Canada Inc. | Unsaturated hydroxyalkylquinoline acids as leukotriene antagonists |
| EP0737186B1 | Dec 22, 1994 | Aug 19, 1998 | Merck & Co., Inc. | Process for the preparation of leukotriene antagonists |
| US2985589 | May 22, 1957 | May 23, 1961 | Universal Oil Prod Co | Continuous sorption process employing fixed bed of sorbent and moving inlets and outlets |
| US5156736 | May 7, 1991 | Oct 20, 1992 | Schoenrock Karlheinz W R | Simulated moving bed apparatus using a single sorbent bed for separating components from a fluid stream |
| US5523477 | Jan 23, 1995 | Jun 4, 1996 | Merck & Co., Inc. | Reacting 1,1-cyclopropanedimethanol with dialkyl sulfite in presence of acid or base to form cyclic sulfite, removing alcohol reaction by-product |
| US5565473 | Feb 23, 1995 | Oct 15, 1996 | Merck Frosst Canada, Inc. | Useful as anti-asthmatic, anti-allergic, anti-inflammatory and cytoprotective agents; montelukast and its sodium salt |
PATENT CITATIONS
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2006008751A2 * | Jul 19, 2004 | Jan 26, 2006 | Satyanarayana Chava | Process for the preparation of montelukast and its salts |
| WO2006043846A1 * | Oct 21, 2005 | Apr 27, 2006 | Inst Farmaceutyczny | Salt of montelukast with tert.-butylamine |
| WO2007072114A1 * | Jan 16, 2006 | Jun 28, 2007 | Harmander Pal Singh Chawla | An improved process for the manufacture of montelukast sodium |
| WO2007107297A1 * | Mar 15, 2007 | Sep 27, 2007 | Synthon Bv | Montelukast amantadine salt |
| US20050107612 * | Dec 30, 2003 | May 19, 2005 | Dr. Reddy's Laboratories Limited | Process for preparation of montelukast and its salts |
NON-PATENT CITATIONS
| Reference | ||
|---|---|---|
| 1 | * | "An improved process to obtain Montelukast sodium" RESEARCH DISCLOSURE, MASON PUBLICATIONS, HAMPSHIRE, GB, vol. 521, no. 2, 1 September 2007 (2007-09-01), page 908, XP007137576 ISSN: 0374-4353 |
| 2 | * | "Piperazine salts of Montelukast, a new efficient method of purification" IP.COM JOURNAL, IP.COM INC., WEST HENRIETTA, NY, US, 29 November 2007 (2007-11-29), XP013122974 ISSN: 1533-0001 |
| 3 | * | AL OMARI ET AL: "Effect of light and heat on the stability of montelukast in solution and in its solid state" JOURNAL OF PHARMACEUTICAL AND BIOMEDICAL ANALYSIS, NEW YORK, NY, US, vol. 45, no. 3, 19 October 2007 (2007-10-19), pages 465-471, XP022306740 ISSN: 0731-7085 |
| 4 | * | DUFRESNE C ET AL: "Synthesis of montelukast (MK-0476) metabolic oxidation products" JOURNAL OF ORGANIC CHEMISTRY, AMERICAN CHEMICAL SOCIETY, EASTON.; US, vol. 61, no. 24, 1 January 1996 (1996-01-01), pages 8518-8525, XP002284162 ISSN: 0022-3263 |
| 5 | * | GRAUL L ET AL: "Montelukast sodium, MK-476, MK-0476, L-706631, Singulair" DRUGS OF THE FUTURE, PROUS SCIENCE, ES, vol. 22, no. 10, 1 January 1997 (1997-01-01), page 1103, XP008082254 ISSN: 0377-8282 |
| 6 | * | NELSON ERIC D ET AL: "Evaluation of solution oxygenation requirements for azonitrile-based oxidative forced degradation studies of pharmaceutical compounds." July 2006 (2006-07), JOURNAL OF PHARMACEUTICAL SCIENCES JUL 2006, VOL. 95, NR. 7, PAGE(S) 1527 - 1539 , XP002563008 ISSN: 0022-3549 compound 4 |
| 7 | * | SMITH GLENN A ET AL: "An automated method for the determination of montelukast in human plasma using dual-column HPLC analysis and peak height summation of the parent compound and its photodegradation product." September 2004 (2004-09), PHARMACEUTICAL RESEARCH SEP 2004, VOL. 21, NR. 9, PAGE(S) 1539 - 1544 , XP002563007 ISSN: 0724-8741 page 1539 - page 1544; example 2 |
| WO2011061545A1 * | Nov 20, 2010 | May 26, 2011 | Generics [Uk] Limited | Hplc method for analyzing vorinostat |
| WO2012077123A1 * | May 12, 2011 | Jun 14, 2012 | Arch Pharmalabs Limited | Purification of montelukast using a simulated moving bed |
| WO2014034203A1 * | May 28, 2013 | Mar 6, 2014 | Dai Nippon Printing Co., Ltd. | Method for producing high-purity montelukast |
| CN102060762A * | Jan 28, 2011 | May 18, 2011 | 海南美大制药有限公司 | Montelukast compound and new preparation method thereof |
| CN102060762B | Jan 28, 2011 | May 29, 2013 | 海南美大制药有限公司 | Montelukast compound and new preparation method thereof |
| US8471030 | Dec 6, 2010 | Jun 25, 2013 | Orochem Technologies Inc. | Purification of montelukast using simulated moving bed |
| US8754129 | Nov 25, 2009 | Jun 17, 2014 | Generics [Uk] Limited | Crystalline vorinostat form VI |
2 PRANLUKAST


PRANLUKAST
Antiasthmatic.
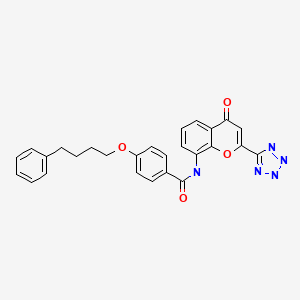
- Benzamide, N-(4-oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl)-4-(4-phenylbutoxy)-
- N-(4-Oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl)-p-(4-phenylbutoxy)benzamide
- 4-Oxo-8-(4-(4-phenylbutoxy)benzoylamino)-2-(tetrazol-5-yl)-4H-1-benzopyran
- N-(4-Oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl)-p-(4-phenylbutoxy)benzamide
| Launched – 1995 japan |
hemihydrate, 103177-37-3 anhydrous, 103180-28-5 (monosodium salt)
150821-03-7, C27 H23 N5 O4 . H2O, 499.5179
Ono-1078
Ono-RS-411
RS-411
SB-205312
Ono-1070 (monosodium salt)
Ono-RS-411
RS-411
SB-205312
Ono-1070 (monosodium salt)
Ultair; Ono-1078; HY-B0290;
- Azlaire
- CCN 00401
- ONO 1078
- ONO-1078
- ONO-RS 411
- Pranlukast
- RS 411
- SB 205312
- UNII-TB8Z891092
N-[4-Oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl]-4-(4-phenylbutoxy)benzamide hemihydrate
Ono (Originator)Schering-Plough (Licensee)

This is described in............
J Med Chem 1988, 31(1): 84,
WO 2010002075,
Synth Commun 1997, 27(6): 1065,
WO 1994012492
Leukotriene antagonist.
Prepn: M. Toda et al., EP 173516; eidem, US 4780469 (1986, 1988 both to Ono);
H. Nakai et al., J. Med. Chem. 31, 84 (1988).
Pharmacology: T. Obata et al., Adv. Prostaglandin Thromboxane Leukotriene Res. 15, 229 (1985); idem et al., ibid. 17,540 (1987).
Clinical evaluations in asthma: Y. Taniguchi et al., J. Allergy Clin. Immunol. 92, 507 (1993); H. Yamamoto et al. Am. J. Respir. Crit. Care Med. 150, 254 (1994).
AU 8546462; EP 0173516; JP 8650977; US 4780469; US 4939141
Pranlukast is a cysteinyl leukotriene receptor-1 antagonist. It antagonizes or reduces bronchospasm caused, principally in asthmatics, by an allergic reaction to accidentally or inadvertently encountered allergens.
 Pranlukast
Pranlukast
Pranlukast is a cysteinyl leukotriene receptor-1 antagonist. This drug works similarly to Merck & Co.'s Singulair (montelukast). It is widely used in Japan.
Medications of this class, which go under a variety of names according to whether one looks at the American, British or European system of nomenclature, have as their primary function the antagonism of bronchospasm caused, principally in asthmatics, by an allergic reaction to accidentally or inadvertently encountered allergens.
Medications of this group are normally used as an adjunct to the standard therapy of inhaled steroids with inhaled long- and/or short-acting beta-agonists. There are several similar medications in the group; all appear to be equally effective.
Pranlukast hydrate is a leukotriene CysLT1 (LTD4) and CysLT2 (LTC4) antagonist first launched in Japan in 1995 as capsules for the oral treatment of bronchial asthma and allergic rhinitis. A dry syrup formulation of pranlukast for the treatment of asthma was approved in Japan in 1999. In April 2011, Ono filed a regulatory application in Japan seeking approval of the compound for the treatment of allergic rhinitis in pediatric patients. In December 2011, approval was obtained for this indication and launch took place immediately.
In terms of clinical development, Ono had been evaluating the drug in phase III for the treatment of sinusitis; however, in 2008 the compound was discontinued for this indication when the compound failed to demostrate the expected efficacy in the phase III studies. In March 2006, Ono discontinued development of the compound for the oral treatment of chronic obstructive pulmonary disease (COPD) based on results which suggested no evidence of efficacy. In 2000, Ono signed a license agreement with Schering-Plough to develop and market pranlukast hydrate in Latin America.
Pranlukast hemihydrate
Also known as: RTR-006167; 150821-03-7..............http://chem.sis.nlm.nih.gov/chemidplus/rn/150821-03-7
- Benzamide, N-(4-oxo-2-(1H-tetrazol-5-yl)-4H-1-benzopyran-8-yl)-4-(4-phenylbutoxy)-, hydrate (2:1)
4-Oxo-8-[(4-phenylbutoxy)benzoylamino]-2-(tetrazol-5-yl)-4H-1-benzopyran · 1/2 hydrate (common name: pranlukast, hereinafter referred to as "pranlukast" in the specification including the claims) represented by formula:

is a compound having a potential antagonistic action against leucotriene C4(LTC4) and leucotriene D4 (LTD4) and is expected as a treating agent for allergic bronchial or pulmonary diseases, allergic shock, and various allergic inflammatory diseases.

 | |
| Systematic (IUPAC) name | |
|---|---|
| N-[4-oxo-2-(1H-tetrazol-5-yl)-4H-chromen-7-yl]-4-(4-phenylbutoxy)benzamide | |
| Clinical data | |
| AHFS/Drugs.com | International Drug Names |
| Legal status |
?
|
| Routes | Oral |
| Pharmacokinetic data | |
| Metabolism | Hepatic (mainly CYP3A4)[1] |
| Half-life | 1.5 hours[1] |
| Identifiers | |
| CAS number | 103177-37-3 |
| ATC code | R03DC02 |
| PubChem | CID 4887 |
| DrugBank | DB01411 |
| ChemSpider | 4718 |
| UNII | TB8Z891092 |
| ChEMBL | CHEMBL21333 |
| Chemical data | |
| Formula | C27H23N5O4 |
| Mol. mass | 481.503 g/mol |
Toda synthetic complete with 3 – nitro-2 – hydroxyphenyl ko one for raw materials, ni ko with oxalic ester Claisen condensation occurs, and then heated to reflux for cyclization to construct benzo pyran ring; dehydrated by an amide synthesized ring cyano group, the cyano compound and then with sodium azide tetrazole synthesis. The nitro group on the compound in 5% Pd / C catalyzed hydrogenation of amino acid reacted with the compound Pranlukast held. This method directly using 4 – (4 – phenyl-butoxy)-benzoic acid reaction. Synthetic route is as follows:
[0006]

[0007]

[0008] ② Robert Graham and routes are routes to I-bromo-butane as a raw material, were used as a palladium catalyst, ligand compound formylation carbonylation reactions and condensation of potassium tert-butoxide, closed dehydration under acidic conditions benzopyran ring method. Synthetic route is as follows:
[0009] Robert routes:
[0010]

[0011] Graham route:
[0012]

[0013] The two synthetic routes are not disclosed in the I-Bromo butane feedstock pathway.
[0014] ③ Masayohi 2_ cyano synthetic route to a benzopyran derivative and hydrogen sulfide gas in the base-catalyzed addition reaction of 2 – thiocarbamoylbenzothiazol and pyran derivatives, and then were reacted with anhydrous hydrazine group hydrazone, with sodium nitrite under acidic conditions nitrosation reaction occurs tetrazole ring. Synthetic route is as follows:
[0015]

[0016] The materials used are not mentioned route synthesis method, it is only reflected in the improvement of the synthesis of the tetrazole ring.
[0017] ④ Giles, Hideki and Hayler are tetrazole substituent on the increase, making it easier condensation reaction, but the synthesis of substituted on the nitrogen with tetrazole difficult, and ultimately elimination reaction of lithium used tetrahydro aluminum and other hazardous reagents, is not easy to Eri industrialization. Reaction scheme is as follows:
[0018]

[0019] ⑤ Lee NK with 4_ (4_ Phenylbutoxy) benzonitrile and 2_ hydroxy _3_ iodobenzene ko 1H_4_ thiazolyl ketone and ester ko _5_ acid, concentrated sulfuric acid catalyzed cyclization iodide copper and potassium phosphate removal under the action of hydrogen iodide get Pranlukast held. Reaction scheme is as follows:

[0021] does not mention the route starting 4 – (4 – phenyl-butoxy)-benzonitrile synthesis method, while two – hydroxy – 3 – Synthesis of iodobenzene ko difficult one.

The synthesis method comprises the following steps: a. 4 – Synthesis of chlorobutanol THF was added concentrated hydrochloric acid, feeding the mass ratio of I: I. 389 ~ 5. 556,45-80 ° C was stirred for 5-18h, cooled, extracted with methylene chloride, removal of the solvent, distillation under reduced pressure to give 4 – chlorobutanol; b. 4 – phenyl butanol take benzene, aluminum chloride mixture ,0-25 ° C solution of 4 – chlorobutanol, reaction 5 -10h then poured into ice-water, a liquid, in addition to homogeneous solution U, distillation under reduced pressure, and the resulting colorless transparent liquid that is, 4 – phenyl butanol; c. I-bromo-4 – phenyl butane synthesis of 4 – phenyl butanol 40% hydrobromic acid mixture, feeding the mass ratio of I: 2. 857 ~ 11. 428, heat refluxing, cooling, liquid separation, the organic solvent divided by distillation under reduced pressure to give I-bromo-4 – phenyl butane; d. Synthesis of methyl p-hydroxybenzoate take-hydroxybenzoic acid and methanol, concentrated sulfuric acid and refluxed for 5-20h spin methanol, poured into cold water to precipitate a white solid which was filtered and dried to give the hydroxy benzoate; e. 4 – (4 – phenyl-butoxy)-benzoic acid methyl ester _ take I-bromo-4 – phenyl butane,
DMF, toluene, methyl p-hydroxybenzoate and potassium carbonate, a reflux 5 ~ 20h, cooling water, extracted with toluene, light yellow liquid rotary evaporation, recrystallization, and the resulting white solid, that is, 4 – (4 – phenyl-butoxy) – benzoic acid methyl ester; f. 4 – (4 – phenyl-butoxy yl) – benzoic acid taken 4 – (4 – phenyl-butoxy) – benzoic acid methyl ester, 15% NaOH solution was refluxed for I ~ 5h, cooled, acidified, filtered and dried to give 4 – (4 – phenylbutyrate oxy) – benzoic acid; g. sprinkle bromophenyl acetic acid ester molar ratio Preparation of I: I ~ I. 5: O. I ~ I of bromophenol, acetic anhydride, pyridine feeding, reflux 3 ~ 10h, distilled pyridine, acetic acid and excess acetic anhydride distilled under reduced pressure to give the acetic acid esters bromophenol; h. 5 – bromo-2 – Preparation of light taken acetophenone molar ratio of I: I ~ 5: I of acetic acid bromophenol esters, aluminum chloride, tetrachlorethylene for feeding, reflux O. 5 ~ 5. 5h, cooled, the reaction solution was poured into 5% hydrochloric acid and extracted with methylene chloride, the solvent evaporated under reduced pressure, to obtain a gray crystalline 5 – bromo-2 – Light acetophenone; i. 5 – bromo-3 – nitro-2 – Preparation of light acetophenone take 5 – bromo-2 – Light acetophenone, carbon tetrachloride, 50 ~ 90 ° C is added dropwise nitric acid, reflux I ~ 4h, cooled, filtered, and the resulting yellow solid which is 5 – bromo-3 – nitro-2 – hydroxyacetophenone; j. 3 – amino-2 – Light benzene ethanone Preparation of 5 – bromo-3 – nitro-2 – hydroxyacetophenone, 5% Pd / C, methylene chloride, methanol, concentrated hydrochloric acid, water, hydrogenation; the end of the reaction mixture was filtered, the filtrate was The solvent was removed, neutralized with sodium bicarbonate, and the resulting yellow solid ginger i.e., 3 – amino-2 – hydroxyacetophenone; k. 3 – [4 - (4 - phenyl-butoxy)-benzoyl amino] -2 _ light base Preparation of acetophenone 4 – (4 – phenyl-butoxy)-benzoic acid, toluene, DMF, 45 ~ 105 ° C was added dropwise SOCl2, 30min the reaction liquid droplets to the 3 – amino-2 – hydroxyphenyl toluene solution of ethyl ketone, the reaction 3 ~ 10h, cooled, neutralized with dilute hydrochloric acid, extracted with toluene, rotary evaporation, and the resulting pale yellow crystals is 3 – [4 - (4_ phenylbutoxy) benzamido] 2_-hydroxyacetophenone; I. 2 – [4 - (4 - phenyl-butoxy)-benzoyl amino] -6 – [l, 3 - dioxo-3 - ethoxycarbonyl-propyl] phenol synthetic sodium, THF, 3 – [4 - (4 - phenyl-butoxy)-benzoyl amino]-2 – hydroxyacetophenone, diethyl oxalate 4 ~ IOh After stirring the reaction was poured into dilute hydrochloric acid to precipitate the yellow solid which was filtered, and the resulting product, i.e. 2 – [4 - (4_ phenylbutoxy) benzamido] _6_ [1,3 - dioxo-3 - ethoxy propyl intended yl] phenyl discretion ·; m. 4 – oxo-8 – [4 - (4 - phenyl-butoxy)-benzoyl amino]-2 – ethoxycarbonyl-4H-benzopyran take 2 – [4 - (4 - phenyl-butoxy yl) benzoyl amino] -6 – [l, 3 - dioxo-3 - ethoxycarbonyl-propyl] phenol, THF, force mouth heat, the addition of concentrated hydrochloric acid, refluxed for 8 ~ 15h, cooled, filtered, and the resulting white solid,
that is, 4 – oxo-8 – [4 - (4 - phenyl-butoxy)-benzoyl amino]-2 – ethoxycarbonyl-4H-benzopyran; η. 4 – oxo-8 – [ 4 - (4 - phenyl-butoxy)-benzoyl amino] -2 – amino-carbonyl-4Η-benzopyran synthesis take four – oxo-8 – [4 - (4 - phenyl-butoxy)-benzoyl amino] -2 – ethoxycarbonyl-4Η-benzopyran was dissolved in DMF,
and leads to dry ammonia gas, the reaction solution changed from yellow to red, the reaction solution was poured into cold water, adjusted to acidic, and filtered to give the product 4 – oxo-8 – [4 - (4 - phenyl-butoxy)-benzoyl amino] -2 – amino-carbonyl-4Η-benzopyran; P. 4 – oxo-8 – [4 - (4 - phenylbutoxy) benzamido] -2 – cyano-4Η-benzopyran take DMF, S0C12, 4 – oxo-8 – [4 - (4 - phenyl-butoxy)-benzoic amido] _2_ aminocarbonyl-4H-benzopyran, O ~ 15 ° C under stirring for 2 ~ IOh poured into cold water, filtered, and the resulting white solid that is, 4 – oxo-8 – [4 - (4 - phenylbutoxy) benzamido] -2 – cyano-4H-benzopyran; q. Synthesis of pranlukast take four – oxo-8 – [4 - (4 - phenyl-butoxy) benzoyl amino]-2_ cyano-4H-benzopyran, ammonium chloride, sodium azide, DMF, heating I ~ 8h then poured into ice-water, dilute hydrochloric acid, filtered, and the resulting white solid that the final product Pranlukast.

The reaction of ethyl 8-nitro-4-oxo-1-benzopyran-2-carboxylate (I) with ammonia in methanol gives the corresponding amide (II), which is dehydrated with POCl3 yielding 2-cyano-8-nitro-1-benzopyran-4-one (III). The cyclization of (III) with sodium azide by means of pyridinium chloride in hot DMF affords 8-nitro-2-(tetrazol-5-yl)-1-benzopyran-4-one (IV), which is hydrogenated with H2 over Pd/C in methanol – HCl giving 8-amino-2-(tetrazol-5-yl)-1-benzopyran-4-one (V). Finally, this compound is condensed with 4-(4-phenylbutoxy)benzoic acid (VI) by means of oxalyl chloride in dichloromethane-pyridine\
 .............................
.............................
Synthetic routes

The reaction of ethyl 8-nitro-4-oxo-1-benzopyran-2-carboxylate (I) with ammonia in methanol gives the corresponding amide (II), which is dehydrated with POCl3 yielding 2-cyano-8-nitro-1- benzopyran-4-one (III). The cyclization of (III) with sodium azide by means of pyridinium chloride in hot DMF affords 8-nitro-2- (tetrazol-5-yl) -1-benzopyran-4-one (IV ), which is hydrogenated with H2 over Pd / C in methanol -. HCl giving 8-amino-2- (tetrazol-5-yl) -1-benzopyran-4-one (V) Finally, this compound is condensed with 4- (4-phenylbutoxy) benzoic acid (VI) by means of oxalyl chloride in dichloromethane-pyridine.
..........................................
PATENT
Example 1: Synthesis of pranlukast
To 10 g of N-(4-oxo-2-(l-trityl-lH-tetrazol-5-yl)-4H-chromen-8-yl)-4-(4- phenylbutoxy) benzamide (Pharmacostech) was added 100 ml of methanol, and 10 g of a resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-B gel type,
Mitsubishi Chemical Co.) was added to the reaction mixture, followed by refluxing for
5 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution and stirred for 1 hour at room temperature. Then, the solid formed was filtered out, dried, and left for 5 hours at room temperature to give 6.32 g (yield:
95%) of the standard compound represented by the following Formula 5: melting point, 231-2330C (decomposed); 1H-NMR (DMSOd6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m,2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0
(m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
 Example 2: Synthesis of pranlukastOne hundred ml of methanol was added to 10 g of N-(4-oxo-2-(l-trityl-lH- tetrazol-5-yl)-4H-chromen-8-yl)-4-(4-phenylbutoxy) benzamide (Pharmacostech), then 10 g of resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-10 gel type, Mitsubishi Chemical Co.) was added to the reaction mixture, followed by refluxing for 6 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution and stirred for 1 hour at room temperature. Then the solid formed was filtered out, dried, and left for 5 hours at room temperature to obtain 6.18 g (yield rate: 93%) of the standard compound represent by Formula 5: melting point, 231- 233°C (decomposed); 1H-NMR (DMSOd6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
Example 2: Synthesis of pranlukastOne hundred ml of methanol was added to 10 g of N-(4-oxo-2-(l-trityl-lH- tetrazol-5-yl)-4H-chromen-8-yl)-4-(4-phenylbutoxy) benzamide (Pharmacostech), then 10 g of resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-10 gel type, Mitsubishi Chemical Co.) was added to the reaction mixture, followed by refluxing for 6 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution and stirred for 1 hour at room temperature. Then the solid formed was filtered out, dried, and left for 5 hours at room temperature to obtain 6.18 g (yield rate: 93%) of the standard compound represent by Formula 5: melting point, 231- 233°C (decomposed); 1H-NMR (DMSOd6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
Example 3: Synthesis of pranlukast
One hundred ml of methanol and 100 ml of methylene chloride (MC) were added to 10 g of N-(4-oxo-2-(l-trityl-lH-tetrazol-5-yl)-4H-chromen-8-yl)-4-(4- phenylbutoxy) benzamide (Pharmacostech), then 10 g of resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-10 gel type) was added to the reaction mixture, followed by refluxing for 12 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution, and stirred for 1 hour at room temperature. Then the solid formed was filtered out, dried, and left for 5 hours at room temperature to obtain 6.18 g (yield rate: 93%) of the standard compound represent by Formula 5: melting point, 231-233°C (decomposed); 1H-NMR (DMSO-d6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
To 10 g of N-(4-oxo-2-(l-trityl-lH-tetrazol-5-yl)-4H-chromen-8-yl)-4-(4- phenylbutoxy) benzamide (Pharmacostech) was added 100 ml of methanol, and 10 g of a resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-B gel type,
Mitsubishi Chemical Co.) was added to the reaction mixture, followed by refluxing for
5 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution and stirred for 1 hour at room temperature. Then, the solid formed was filtered out, dried, and left for 5 hours at room temperature to give 6.32 g (yield:
95%) of the standard compound represented by the following Formula 5: melting point, 231-2330C (decomposed); 1H-NMR (DMSOd6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m,2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0
(m, 2H), 8.3 (t, IH), 10.0 (bs, IH).

Example 3: Synthesis of pranlukast
One hundred ml of methanol and 100 ml of methylene chloride (MC) were added to 10 g of N-(4-oxo-2-(l-trityl-lH-tetrazol-5-yl)-4H-chromen-8-yl)-4-(4- phenylbutoxy) benzamide (Pharmacostech), then 10 g of resin pre-treated with hydrochloric acid of pH 2-3 (TRILITE SCR-10 gel type) was added to the reaction mixture, followed by refluxing for 12 hours. The solid components were filtered out from the reaction mixture and washed with 100 ml of methanol. The filter-in solution was subject to vacuum distillation to obtain a solid substance and the solid was dissolved in 50 ml of dimethyl acetamide (DMAC). Afterwards, 200 ml of aqueous solution was added to the DMAC solution, and stirred for 1 hour at room temperature. Then the solid formed was filtered out, dried, and left for 5 hours at room temperature to obtain 6.18 g (yield rate: 93%) of the standard compound represent by Formula 5: melting point, 231-233°C (decomposed); 1H-NMR (DMSO-d6, 300 MHz), δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
................................................................
Pranlukast and its hydrates come into the market as a capsule of Onon® Cap. (112.5 mg pranlukast hydrates/capsule, Dong-A Pharmaceutical).
 The conventional method for preparing pranlukast was disclosed in US Pat. No. 5,587,483 and pranlukart is prepared by the following reaction formula I.
The conventional method for preparing pranlukast was disclosed in US Pat. No. 5,587,483 and pranlukart is prepared by the following reaction formula I.
Reaction Formula I
 As described in the reaction formula I, the acid chloride represented by formula 11 is obtained by reacting the benzoic derivative of formula 10 with the thionyl chloride. The resulting compound is reacted with the compound represented by formula 4. The compound (n = 4) represented by formula 5 is reacted with the tetrazol derivative represented by formula 6 to introduce tetrazol group and then benzopyran ring is formed, preparing pranlukast. However, the preparation method according to the reaction formula I has quite a few problems: (a) difficult manipulation due to utilizing excess amounts of toxic thionyl chlorides around a reflux temperature when the acid chloride represented by formula 11 is obtained by reacting the benzoic derivative of formula 10 with the thionyl chloride;
As described in the reaction formula I, the acid chloride represented by formula 11 is obtained by reacting the benzoic derivative of formula 10 with the thionyl chloride. The resulting compound is reacted with the compound represented by formula 4. The compound (n = 4) represented by formula 5 is reacted with the tetrazol derivative represented by formula 6 to introduce tetrazol group and then benzopyran ring is formed, preparing pranlukast. However, the preparation method according to the reaction formula I has quite a few problems: (a) difficult manipulation due to utilizing excess amounts of toxic thionyl chlorides around a reflux temperature when the acid chloride represented by formula 11 is obtained by reacting the benzoic derivative of formula 10 with the thionyl chloride;
(b) hard elimination of thionyl chlorides toxic in a body after terminating the reactions; (c) requirement of base in an equivalent ratio of above 4 to collect the compound represented by formula 7; (d) unsuitability of massive production in a economical area because the compound is modified into a form of natrium salt and then purified for removal of contaminants after preparing pranlukart.
On the other hand, as described in the following reaction formula II in US Pat. No. 5,874,593, nitril compounds of formula 8 are reacted with hydrazine to prepare amidrazone compounds of formula 9a and 9b, and then pranlukart is fabricated by performing a tetrazol ring reaction using nitrous acids.
Reaction Formula II
 However, the preparation method according to the reaction formula II has also the following difficulties: (a) it is difficult to perform the method due to utilizing excess amounts of toxic thionyl chlorides around a reflux temperature to obtain the acid chloride derivative in the preparation of the compounds represented by formula 8; (b) it is very difficult and toxic in body to eliminate thionyl chlorides after terminating the reactions; (c) it is not easy to massively produce the compounds of interest in an industrial-scale because much hydrazine toxic in body and nitrogen oxides harmful in environment are generated and unstable nitrous acids are used during the reactions.
However, the preparation method according to the reaction formula II has also the following difficulties: (a) it is difficult to perform the method due to utilizing excess amounts of toxic thionyl chlorides around a reflux temperature to obtain the acid chloride derivative in the preparation of the compounds represented by formula 8; (b) it is very difficult and toxic in body to eliminate thionyl chlorides after terminating the reactions; (c) it is not easy to massively produce the compounds of interest in an industrial-scale because much hydrazine toxic in body and nitrogen oxides harmful in environment are generated and unstable nitrous acids are used during the reactions.
Likewise, US Pat. No. 5,874,593, as described in the following reaction formula III, discloses that benzoic derivatives of formula 10' are reacted with oxalyl chlorides to isolate acid chlorides represented by formula 11', and the resulting acid chlorides are reacted with benzopyran amine derivatives containing tetrazol of formula 12, producing various derivatives containing pranlukart.
Reaction Formula III
 ( I D' ] (H ' )
( I D' ] (H ' )
 Oxalyl chlorides are massively used because the preparation method according to the reaction formula III is very expensive cost and has highly hygroscopic characteristics. In addition, the method has to be carried out under violent conditions that the temperature is increased up to around reflux temperature using 1,2- dichloroethanol as a solvent and further reacted for 1 hr. It is also difficult to remove harmful carbon monoxide and chlorine gases massively generated in elimination of oxalyl chloride after terminating the reactions, and it is not feasible to be applied into an industrial mass-production because the reaction is carried out under conditions of anhydrous and inactive gases
Oxalyl chlorides are massively used because the preparation method according to the reaction formula III is very expensive cost and has highly hygroscopic characteristics. In addition, the method has to be carried out under violent conditions that the temperature is increased up to around reflux temperature using 1,2- dichloroethanol as a solvent and further reacted for 1 hr. It is also difficult to remove harmful carbon monoxide and chlorine gases massively generated in elimination of oxalyl chloride after terminating the reactions, and it is not feasible to be applied into an industrial mass-production because the reaction is carried out under conditions of anhydrous and inactive gases

Reaction Formula I

(b) hard elimination of thionyl chlorides toxic in a body after terminating the reactions; (c) requirement of base in an equivalent ratio of above 4 to collect the compound represented by formula 7; (d) unsuitability of massive production in a economical area because the compound is modified into a form of natrium salt and then purified for removal of contaminants after preparing pranlukart.
On the other hand, as described in the following reaction formula II in US Pat. No. 5,874,593, nitril compounds of formula 8 are reacted with hydrazine to prepare amidrazone compounds of formula 9a and 9b, and then pranlukart is fabricated by performing a tetrazol ring reaction using nitrous acids.
Reaction Formula II

Likewise, US Pat. No. 5,874,593, as described in the following reaction formula III, discloses that benzoic derivatives of formula 10' are reacted with oxalyl chlorides to isolate acid chlorides represented by formula 11', and the resulting acid chlorides are reacted with benzopyran amine derivatives containing tetrazol of formula 12, producing various derivatives containing pranlukart.
Reaction Formula III

EXAMPLE 1: Preparation of Pranlukart Hemihydrates 4-(4-phenylbutoxy)benzoic acid (29.1 g; 1.1 equivalent ratio; prepared according to the method disclosed in US Pat. No. 4,780,469) was dissolved in 80 ml dimethylacetamide (DMAC, Aldrich) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio, Aldrich) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H- 1-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio; prepared according to the method disclosed in US Pat. No. 4,780,469) and triethylamine (TEA, 10.1 g, 1 equivalent ratio, Aldrich) dissolved in 80 ml dimethylacetamide (DMAC, Aldrich) was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 250C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 47.0 g pranlukart hemihydrates (yield rate: 98%): melting point 231-233°C (decomposition); 1H-NMR (DMSO-d6, 300 MHz) δ 1.9 (m, 4H), 2,7 (m, 2H), 4.0 (t, 2H), 7.0 (s, 2H), 7.1 (s, IH), 7.2-7.3 (m, 5H), 7.6 (t, IH), 7.9 (t, IH), 8.0 (m, 2H), 8.3 (t, IH), 10.0 (bs, IH).
EXAMPLE 2: Preparation of Pranlukart Hemihydrates - Substitution of the Chlorinating Agent
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then oxalyl chloride (15.2 g, 1.2 equivalent ratio, Aldrich) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H- 1-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEA, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 43.3 g pranlukart hemihydrates (yield rate: 92%).
EXAMPLE 3: Preparation of Pranlukart Hemihydrates - Change of Base Condition
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and pyridine (7.9 g, 1 equivalent ratio, Aldrich) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 5 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 45.6 g pranlukart hemihydrates (yield rate: 95%).
EXAMPLE 4: Preparation of Pranlukart Hemihydrates - Change of Reaction Temperature Condition
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at O0C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 500C. The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 500C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 45.6 g pranlukart hemihydrates (yield rate: 95%).
EXAMPLE 5: Preparation of Pranlukart Hemihydrates - Substitution of Reaction Solvent 4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml N-methylpyrrolidine (NMP, Aldrich) at O0C and then thionyl chloride (14.2 g, 1.2 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g; 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml N-methylpyrrolidine (NMP) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 250C. The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C.
The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 43.3 g pranlukart hemihydrates (yield rate: 90%).
EXAMPLE 6: Preparation of Pranlukart Hemihydrates - Equivalent Ratio Change of the Chlorinating Agent
4-(4-phenylbutoxy)benzoic acid (29.1 g, 1.1 equivalent ratio) was dissolved in 80 ml dimethylacetamide (DMAC) at 00C and then thionyl chloride (14.2 g, 1 equivalent ratio) was gradually added to the solution. After the mixture solution was stirred for 10 min at 00C, the mixture of 8-amino-4-oxo-tetrazol-5-yl-4H-l-benzopyran hydrochloride salt (26.7 g, 1 equivalent ratio) and triethylamine (TEZ, 10.1 g, 1 equivalent ratio) dissolved in 80 ml dimethylacetamide (DMAC) solution was slowly added to the mixture solution, and thermally stirred for 4 hrs at 25°C.
The reaction mixture was mixed with 300 ml H2O and stirred for 1 hr at 25°C. The solid material obtained by filtering the solid material produced was washed with 100 ml H2O. 200 ml 50% acetone aqueous solution was added to the solid material and then refluxed for 1 hr. After the reaction mixture was cooled to room temperature, filtered and air-dried, the mixture was kept to stand on air for 5 hrs, obtaining 44.6 g pranlukart hemihydrates (yield rate: 93%).
..........................
J. Med. Chem., 1988, 31 (1), pp 84–91
DOI: 10.1021/jm00396a013
...........................................................
Geen, G.R.; Giles, R.G.; Grinter, T.J.; Hayler, J.D.; et al.
A direct and high yielding route to 2-(5-tetrazolyl) substituted benzopyran-4-ones: Synthesis of pranlukast
Synth Commun 1997, 27(6): 1065
A direct and high yielding route to 2-(5-tetrazolyl) substituted benzopyran-4-ones: Synthesis of pranlukast
Synth Commun 1997, 27(6): 1065
A direct and high yielding route to 2-(5-tetrazolyl)benzopyran-4-ones 1, including pranlukast 1a is described. This involves the Claisen condensation reaction between the relevant hydroxyacetophenone 2 and the ethyl ester of tetrazole-2-carboxylic acid 5 to give the 1,3-diketone 6, which is then cyclised to give the desired benzopyran-4-ones 1.\
........................................................
WO 1994012492
References
- Nakade S, Ueda S, Ohno T, Nakayama K, Miyata Y, Yukawa E, Higuchi S (2006). "Population pharmacokinetics of pranlukast hydrate dry syrup in children with allergic rhinitis and bronchial asthma.". Drug Metab Pharmacokinet 21 (2): 133–9. doi:10.2133/dmpk.21.133. PMID 16702733.
3 ZAFIRLUKAST

ZAFIRLIKAST 
cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate 107753-78-6
Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 (1990);
U. S. Patent No. 4,859,692;
U. S. Patent No. 5,993,859;
Zafirlukast is an oral leukotriene receptor antagonist (LTRA) for the maintenance treatment of asthma, often used in conjunction with an inhaled steroid and/or long-acting bronchodilator. It is available as a tablet and is usually dosed twice daily. Another leukotriene receptor antagonist is montelukast (Singulair), taken once daily. Zileuton (Zyflo), also used in the treatment of asthma via its inhibition of 5-lipoxygenase, is taken four times per day.
Zafirlukast blocks the action of the cysteinyl leukotrienes on the CysLT1 receptors, thus reducing constriction of the airways, build-up of mucus in the lungs andinflammation of the breathing passages.
Zafirlukast is marketed by Astra Zeneca with the brand names Accolate, Accoleit, and Vanticon. It was the first LTRA to be marketed in the USA and is now approved in over 60 countries, including the UK, Japan, Taiwan, Italy, Spain, Canada, Brazil, China and Turkey
Healthy young men who received a single oral 40 mg dose attained peak plasma zafirlukast concentrations that averaged 607 μg/L at 3.4 hours. The elimination half-life ranged from 12 to 20 hours. In another study involving a 20 mg single oral dose in healthy men, the elimination half-life averaged 5.6 hours.[1][2]
A letter was submitted to the FDA by Zeneca Pharmaceuticals on July 22, 1997, notifying them of a change in product labeling that includes the following potential reaction in patients undergoing a dosage reduction of oral steroids who are currently taking zafirlukast:
PRECAUTIONS-Eosinophilic Conditions: The reduction of the oral steroid dose, in some patients on ACCOLATE therapy, has been followed in rare cases by the occurrence of eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy sometimes presenting as Churg–Strauss syndrome, a systemic eosinophilic vasculitis. Although a causal relationship with ACCOLATE has not been established, caution is required when oral steroid reduction is being considered.1
NDA..020547 26/09/1996, ACCOLATE, ASTRAZENECA, 20MG TABLET
| US Patent No | Expirey Date | patent use code |
|---|---|---|
| 5482963 | Jan 9, 2013 | |
| 5612367 | Mar 18, 2014 | U-189 |
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| - | R03DC01 | C 31 H 33 N 3 O 6 S | 575.69 g / mol | 107753-78-6 |
| monohydrate | R03DC01 | C 31 H 33 N 3 O 6 S · H 2 O | 593.70 g / mol | 143052-93-1 |
| calcium (2: 1) | R03DC01 | C 62 H 64 CaN 6 O 12 S 2 | 1189.43 g / mol | 107753-86-6 |
Application
- antihistamine effect
- LTD4-antagonist
Classes of substances
- Benzenesulfonamide (s -imidy), as well as their derivatives
- Esters of carbamic acid
- Cyclopentanes
- Hydroxybenzoic acid amides, and hydroxy acids alkoksibenzoynyh
- Indoles
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), with the chemical name 4(5-cyclopentyloxy-carbonylamino-1-methyl-indol-3ylmethyl)-3-methoxy-N-o-tolylsulfonylbenzamide. The molecular weight of zafirlukast is 575.7 and the structural formula is:
 Zafirlukast, a fine white to pale yellow amorphous powder, is practically insoluble in water. It is slightly soluble in methanol and freely soluble in tetrahydrofuran, dimethylsulfoxide, and acetone.The empirical formula is: C31H33N3O6S
Zafirlukast, a fine white to pale yellow amorphous powder, is practically insoluble in water. It is slightly soluble in methanol and freely soluble in tetrahydrofuran, dimethylsulfoxide, and acetone.The empirical formula is: C31H33N3O6S- Fischer JD, Song MH, Suttle AB, Heizer WD, Burns CB, Vargo DL, Brouwer KL. Comparison of zafirlukast (Accolate) absorption after oral and colonic administration in humans. Pharmaceut. Res. 17: 154-159, 2000.
- Bharathi DV, Naidu A, Jagadeesh B, Laxmi KN, Laxmi PR, Reddy PR, Mullangi R. Development and validation of a sensitive LC-MS/MS method with electrospray ionization for quantitation of zafirlukast, a selective leukotriene antagonist in human plasma: application to a clinical pharmacokinetic study. Biomed. Chromatogr. 22: 645-653, 2008.
- Zafirlukast (U.S. National Library of Medicine)
- Zafirlukast (patient information)

 | |
 | |
| Systematic (IUPAC) name | |
|---|---|
| cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate | |
| Clinical data | |
| Trade names | Accolate |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a697007 |
| Pregnancy cat. | B1 (Australia), B (United States) |
| Legal status | POM (UK) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | Unknown |
| Protein binding | 99% |
| Metabolism | Hepatic (CYP2C9-mediated) |
| Half-life | 10 hours |
| Excretion | Biliary |
| Identifiers | |
| CAS number | 107753-78-6 |
| ATC code | R03DC01 |
| PubChem | CID 5717 |
| IUPHAR ligand | 3322 |
| DrugBank | DB00549 |
| ChemSpider | 5515 |
| UNII | XZ629S5L50 |
| KEGG | D00411 |
| ChEBI | CHEBI:10100 |
| ChEMBL | CHEMBL603 |
| Chemical data | |
| Formula | C31H33N3O6S |
| Mol. mass | 575.676 g/mol |
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| United Kingdom | Akkolat | AstraZeneca |
| Italy | Akkoleit | - "- |
| Zafirst | Chiesi | |
| Japan | Akkolat | AstraZeneca |
| USA | - "- | Zeneca |
| Ukraine | No | No |
Formulations
- Tablets of 20 mg, 40 mg
Zafirlukast, cyclopentyl 3 - [2-methoxy-4- [(o-tolylsulfonyl)carbamoyl]- benzyl]-l-methyIindole-5-carbamate, having the formula:


Alkyl (l-alkylindol-3-ylmethyl)benzoates of formula [lb] are useful as chemical intermediates in the pharmaceutical industry.

These compounds may be obtained by a process described in J. Med. Chem., v. 33, 1781 (1990) and U. S. Patent 4,859,692. This process comprises the steps of:
(a) reacting an alkyl (halomethyl)benzoate of formula [2] with an equivalent amount of an indole of formula [3]

(b) isolating the alkyl (indol-3-ylmethyl)benzoates of formula [4] from the reaction mixture obtained in step (a) above,
(c) reacting the compound [4] with an alkylating agent of formula [6],


Formula (I) compound for the synthesis of an important intermediate of zafirlukast.Reported in the patent EP199543 synthesized compound (I) of the conventional method, the following formula:

(A) (I)
In this method, Intermediate A and 5 - nitro-indole silver oxide in the presence of a catalyst, for docking composite formula (I) compound. Reported only 45% of the reaction yield, the reaction is difficult to complete the reaction and post-treatment using chromatographic methods, resulting in product purification more difficult. And the use of more expensive silver oxide catalysts, high cost.
W00246153 reported a catalyst for the above reaction to zinc bromide, Compound (I), after treatment of the compound (I) with sodium hydroxide hydrolysis of the intermediate (B), separating the product and raw materials purification products.

The method reported in the literature a yield of 60%, but the actual operation is repeated only about 30% yield, and the operation is complicated, cumbersome and costly.
zaafirlukast is a selective and competitive receptor antagonist of leukotriene D4 and E4 (LTD4 and LTE4), components of slow-reacting substance of anaphylaxis (SRSA). Cysteinyl leukotriene production and receptor occupation have been correlated with the pathophysiology of asthma, including airway edema, smooth muscle constriction, and altered cellular activity associated with the inflammatory process, which contribute to the signs and symptoms of asthma.
The cysteinyl leukotrienes (LTC4 LTD4, LTE4) are the products of arachidonic acid metabolism and are various cells, including mast cells and eosinophills, these eicosinoids bind to cysteinyl leukotriene (CysLT) receptors. The CysLT type-1 (CysLT1) receptor is found in human airway and other pro-inflammatory cells. CysLTs have been correlated with the pathophysiology of asthma.
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), useful for the treatment of asthma and is commercially available in products sold under the brand name ACCOLATE™ as 10 and 20 mg tablets for oral administration. ACCOLATE™ is indicated for the prophylaxis and treatment of asthma in adults and children 5 years of age and older.
ACCOLATE™ film coated tablets contain amorphous zafirlukast as the active ingredient and the excipients croscarmellose sodium, lactose, magnesium stearate, microcrystalline cellulose, povidone, hypromellose, and titanium dioxide.
The greatest prevalence of asthma is in preschool children; however, the clinical utility of asthma therapy for this age group is limited by a narrow therapeutic index, long-term tolerability, and frequency and/or difficulty of administration. Asthma treatment requires an immediate perceivable effect. Inhalation therapy is a very common therapy prescribed for young children; inhalation therapy has the disadvantage of high dose variability.

........................
Process for the preparation of zafirlukastUS 20040186300 A1

In comparison, the known process for the preparation of zafirlukast described in J. Med. Chem., v. 33, 1781 (1990) and U.S. Pat. No. 4,859,692 involves separation steps, e.g. column chromatography, that are not practical for industrial scale applications. The known process is summarized in Scheme 3:

,..............................................................

An Improved and Scalable Process for Zafirlukast: An Asthma Drug
Research and Development, Integrated Product Development, Dr. Reddy’s Laboratories Ltd., Survey No.’s 42, 45, 46, and 54, Bachupally, Qutubullapur, Ranga Reddy District - 500 072, Andhra Pradesh, India, Institute of Science and Technology, Center for Environmental Science, J.N.T. University, Kukatpally, Hyderabad - 500 072, Andhra Pradesh, India, and Research and Development, Inogent Laboratories Private Limited (A GVK BIO Company), 28A, IDA, Nacharam, Hyderabad - 500 076, India
Org. Process Res. Dev., 2009, 13 (1), pp 67–72
DOI: 10.1021/op800137b
Synthesis pathway
| Synthesis a) |
|---|
      |
| Synthesis of b) |
   |
- Synthesis a)
- US 4,859,692 (ICI; 08/22/1989; GB -prior. 4/17/1985; 17.10.1985).
- EP 199 543 (ICI, Zeneca; appl. 16.4.1986; GB -prior. 4/17/1985).
- Synthesis of b)
- EP 490 649 (ICI, Zeneca; 11.12.1991; GB -prior. 12.12.1990).
- Matassa, G. et al .: J. Med. Chem. (JMCMAR) 33, 1781 (1990).
- Srinivas, K. et al .: Org. Process Res. Dev. (OPRDFK) 8 (6), 952 (2004).

zafirlukast.......{3-[2-Methoxy-4-(toluene-2-sulfonylaminocarbonyl) benzyl]-1-methyl-1H-indol-5-yl} acetic acid cyclopentyl ester.....................................Arie, G.; Genndy, N.; Igor, Z.; Victor, P.; Maxim, S. WO 02/46153 A2, 2002. 

4
4 ibudilast

IBUDILAST
AV-411
KC-404
MN-166
KC-404
MN-166
2-methyl-1-(2-propan-2-ylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one
KYORIN Kyorin Seiyaku Kk……….INNOVATOR
Properties: Crystals from hexane, mp 53.5-54°. Slightly sol in water, freely sol in organic solvents. LD50 i.v. in mice: 260 mg/kg (Irikura, 1973).
Melting point: mp 53.5-54°
Toxicity data: LD50 i.v. in mice: 260 mg/kg (Irikura, 1973)
Therap-Cat: Antiallergic; antiasthmatic; vasodilator (cerebral).

Ibudilast is an anti-inflammatory and neuroprotective oral agent which shows an excellent safety profile at 60 mg/day and provides significantly prolonged time-to-first relapse and attenuated brain volume shrinkage in patients with relapsing-remitting (RR) and/or secondary progressive (SP) multiple sclerosis (MS). Ibudilast is currently in development in the U.S. (codes: AV-411 or MN-166), but is approved for use as an antiinflammatory in Japan.
cas: 50847-11-5, Ibudilastum, Ketas, KC-404, MN-166, Ke Tas
Molecular Formula:C14H18N2O
Molecular Weight:230.30552 g/mol

Ibudilast is a leukotriene antagonist and phosphodiesterase inhibitor which has been commercialized in Japan and other parts of Asia for over 15 years as capsules for the treatment of bronchial asthma and cerebrovascular disturbances. In 2000, the product was launched for the topical treatment of allergic conjunctivitis. At present, the drug is undergoing phase II trials at MediciNova for the oral treatment of multiple sclerosis (MS), for the treatment of medication overuse headache (MOH), for the treatment of diabetic neuropathic pain, for the treatment of methamphetamine addiction and for the treatment of opioid withdrawal.
Early clinical trials are ongoing for the treatment of amyotrophic lateral sclerosis as an adjunt to riluzole.MediciNova and the University of Colorado had been evaluating the product in preclinical studies for the treatment of post-traumatic brain injury; however, no recent development has been reported for this research.Ibudilast exhibits antiplatelet properties mainly by inhibiting phosphodiesterase V (PDE V), which in turn potentiates antiplatelet function of endothelium-derived nitric oxide (NO). The compound has also been shown to suppress hippocampal apoptosis induced by amyloid beta hypoxia.

Ibudilast (development codes: AV-411 or MN-166) is an antiinflammatory drug used mainly in Japan, which acts as aphosphodiesterase inhibitor, inhibiting the PDE-4 subtype to the greatest extent,[1] but also showing significant inhibition of other PDE subtypes.[2][3]
Ibudilast has bronchodilator, vasodilator [4] and neuroprotective effects,[5][6] and is mainly used in the treatment of asthma andstroke.[7] It inhibits platelet aggregation,[8] and may also be useful in the treatment of multiple sclerosis.[9]
Ibudilast crosses the blood–brain barrier and suppresses glial cell activation. This activity has been shown to make ibudilast useful in the treatment of neuropathic pain and it not only enhances analgesia produced by opioid drugs, but also reduces the development oftolerance.[10]

It may have some use reducing methamphetamine addiction.[11]
Avigen has identified the potential of ibudilast (AV-411) for the treatment of neuropathic pain and other neurological indications, including opiate withdrawal. As an inhibitor of glial cells, ibudilast can deactivate these cells which produce various chemicals, including proinflammatory cytokines, in response to nerve damage or viral infection to amplify and maintain pain. Preclinical evaluation to date indicates that it reverses the painful sensory abnormality allodynia in chemotherapy- and trauma-induced neuropathic pain models.
Originator Kyorin and Banyu Pharmaceutical (now MSD KK following the merger of Banyu and Schering-Plough KK in 2010) have been developing ibudilast under a collaborative agreement. MediciNova obtained exclusive, worldwide rights outside of Japan, China, Taiwan and South Korea from Kyorin in October 2004 to develop and commercialize the compound for MS. In 2012, a codevelopment agreement was signed between MediciNova and the University of Colorado for the treatment of post-traumatic brain injury.

Sixteenth revised Japanese Pharmacopoeia chemicals, etc. IBUDILAST Ibudilast C14H18N2O: 230.31 [ 50847-11-5 ] that this product was dried when to quantify, including ibudilast (C14H18N2O) 98.5 ~ 101.0%.


 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| 2-methyl-1-(2-propan-2-ylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one | |
| CLINICAL DATA | |
| AHFS/DRUGS.COM | International Drug Names |
| IDENTIFIERS | |
| CAS NUMBER | 50847-11-5 |
| ATC CODE | R03DC04 |
| PUBCHEM | CID 3671 |
| DRUGBANK | DB05266 |
| CHEMICAL DATA | |
| FORMULA | C14H18N2O |
| MOL. MASS | 230.31 g/mol |
……………………………
EXAMPLE 1 Synthesis of 2-isopropyl-3-is0butyrylpyrazolo[1,5-a] pyridine (KC404) A mixture of 1-amino-Z-methylpyridinium iodide g.), isobutyric anhydride (500 g.) and K CO (81 g.) was refluxed for 8 hr. After cooling, the precipitated crystals were filtered off and water was added to the filtrate, The solution was made basic to pH 11 with K CO’ and extracted with ethyl acetate (1000 ml.). The extract’was washed with water (400 ml.), dried over Na SO and concentrated under reduced pressure. The residue was distilled to give 58 g. of colorless crystalline product, hp, 110- 175 (7.5 mm. Hg). Recrystallization from hexane gave colorless prisms, melting point 53.554.
Analysis- Calcd.: C, 73.01; H, 7.88; N, 12.17 Found: C, 72.86; H, 7.94; N, 12.09
………………………………………..

………………………..
FIG. 6 is a synthetic reaction scheme illustrating one approach for preparing (S)-AV1013; the approach employs chiral chromatography of an N-protected form of the racemate as described in detail in Example 1.
FIG. 7 demonstrates additional reaction schemes for synthesizing (S)-AV1013.


Example 1Synthesis of (S)-2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one hydrochloride
(S)-2-Amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one hydrochloride (also referred to herein as S-AV1013.HCl) was prepared on a preparative scale using two different routes to obtain the intermediate isopropylpyrazolo[1,5-a]pyridine (IPPP). In the first approach (method 1), ibudilast was employed as the starting material to obtain IPPP; an alternate synthetic approach (method 2) employed ibudilast acid as the starting material.
Step 1Method 1Preparation of Isopropylpyrazolo[1,5-a]pyridine (IPPP) from ibudilast

A 5 L 3-neck round-bottom flask was equipped with a mechanical stirrer, thermocouple, heating mantle and a Y-adapter with a nitrogen inlet. The flask was charged with water (350 mL, USP), concentrated sulfuric acid (350 mL) and ibudilast (3-isobutyryl-2-isopropylpyrazolo[1,5-a]pyridine) (140 g, 0.608 mol). The flask was purged with nitrogen, and the mixture was stirred while it was heated to 135° C. An aliquot was removed for HPLC analysis, which showed that all starting material was consumed after 5 hours at 135° C., so the mixture was allowed to cool to room temperature overnight. The mixture was cooled in an ice bath, and water (1400 mL, USP) was added over 10 min, with the temperature maintained below 25° C. With continuous cooling in an ice bath, the mixture was neutralized by adding sodium hydroxide (50% w/w aq., 1150 mL) dropwise, with the temperature maintained below 25° C. Ethyl acetate (250 mL) was added, and the layers were separated. The aqueous layer was washed with ethyl acetate (2×300 mL). The combined ethyl acetate extracts were washed sequentially with 250 mL portions of saturated aqueous sodium bicarbonate and saturated aqueous sodium chloride, then dried over anhydrous sodium sulfate for 30 minutes. Activated carbon (20 g) and silica (60 g) were added and stirred before filtering over a pad of Celite. The filtrate was concentrated under reduced pressure to obtain 96.5 g of IPPP (2-isopropyl-pyrazolo[1,5-a]pyridine, 99% crude yield, 99.6 area % pure by HPLC) as an amber oil.
1H-NMR (CDCl3) δ 1.4 (d, 6H), 3.2 (m, 1H), 6.3 (s, 1H), 6.6 (t, 1H), 7.0 (m, 1H), 7.4 (d, 1H), 8.4 (d, 1H). HPLC: RT=9.1 min (99.6 area %).
………………………………………..
Ibudilast (3-isobutyryl-2-isopropylpyrazolo[l,5-α]pyridine) is a small molecule drug that has been used for many years in Japan and Korea for the treatment of bronchial asthma as well as for treatment of cerebrovascular disorders such as post-stroke dizziness. It is sold in these countries under the tradename, Ketas®. Marketed indications for ibudilast in Japan include its use as a vasodilator, for treating allergy, eye tissue regeneration, ocular disease, and treatment of allergic ophthalmic disease (Thompson Current Drug Reports). Its use in the treatment of both chronic brain infarction (ClinicalTrials.gov) and multiple sclerosis (News.Medical.Net; Pharmaceutical News, 2 Aug 2005) is currently being explored in separate, ongoing clinical trials.
The mechanisms of action of ibudilast have been widely explored. Its role as a non-selective inhibitor of cyclic nucleotide phosphodiesterase (PDE) has been described
(Fujimoto, T., et al., J. of Neuroimmunology, 95 (1999) 35-92). Additionally, ibudilast has been reported to act as an LTD4 antagonist, an anti-inflammatory, a PAF antagonist, and a vasodilatator agent (Thompson Current Drug Reports). Ibudilast is also thought to exert a neuroprotective role in the central nervous system of mammals, presumably via suppression of the activation of glial cells (Mizuno et al. (2004) Neuropharmacology 46: 404-411). New uses for ibudilast continue to be explored.http://www.google.com/patents/WO2007146087A2?cl=en
…………………………
IBUDILAST
Ibudilast is a small molecule drug (molecular weight of 230.3) having the structure shown below.

Ibudilast is also found under ChemBank ID 3227, CAS # 50847-1 1-5, and Beilstein Handbook Reference No. 5-24-03-00396. Its molecular formula corresponds to [Ci4HIgN2O]. Ibudilast is also known by various chemical names which include 2- methyl-l-(2-(l-methylethyI)pyrazolo(l,5-a)pyridin-3-yl)l-propanone; 3-isobutyryl-2- isopropylpyrazolo(l,5-a)pyridine]; and l-(2-isopropyl-pyrazolo[l,5-a]pyridin-3-yl)-2- methyl-propan-1-one. Other synonyms for ibudilast include Ibudilastum (Latin), BRN 0656579, KC-404, and the brand name Ketas®. Ibudilast, as referred to herein, is meant to include any and all pharmaceutically acceptable salt forms thereof, prodrug forms (e.g., the corresponding ketal), and the like, as appropriate for use in its intended formulation for administration.
Ibudilast is a non-selective nucleotide phosphodiesterase (PDE) inhibitor (most active against PDE-3 and PDE-4), and has also been reported to have LTD4 and PAF antagonistic activities. Its profile appears effectively anti-inflammatory and unique in comparison to other PDE inhibitors and anti-inflammatory agents. PDEs catalyze the hydrolysis of the phosphoester bond on the 3 ‘-carbon to yield the corresponding 5′- nucleotide monophosphate. Thus, they regulate the cellular concentrations of cyclic nucleotides. Since extracellular receptors for many hormones and neurotransmitters utilize cyclic nucleotides as second messengers, the PDEs also regulate cellular responses to these extracellular signals. There are at least eight classes of PDEs: Ca2+/calmodul in-dependent PDEs (PDEl); cGMP-stimulated PDEs (PDE2); cGMP- inhibited PDEs (PDE3); cAMP-specific PDEs (PDE4); cGMP-binding PDEs (PDE5); photoreceptor PDEs (PDE6); high affinity, cAMP-specific PDEs (PDE7); and high affinity cGMP-specific PDEs (PDE9).
References
- Huang Z, Liu S, Zhang L, Salem M, Greig GM, Chan CC, Natsumeda Y, Noguchi K. Preferential inhibition of human phosphodiesterase 4 by ibudilast. Life Sciences. 2006 May 1;78(23):2663-8.
- Suzumura A, Ito A, Yoshikawa M, Sawada M. Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS. Brain Research. 1999 Aug 7;837(1-2):203-12.
- Gibson LC, Hastings SF, McPhee I, Clayton RA, Darroch CE, Mackenzie A, Mackenzie FL, Nagasawa M, Stevens PA, Mackenzie SJ. The inhibitory profile of Ibudilast against the human phosphodiesterase enzyme family. European Journal of Pharmacology. 2006 May 24;538(1-3):39-42.
- Kishi Y, Ohta S, Kasuya N, Sakita S, Ashikaga T, Isobe M. Ibudilast: a non-selective PDE inhibitor with multiple actions on blood cells and the vascular wall.Cardiovascular Drug Reviews. 2001 Fall;19(3):215-25.
- Mizuno T, Kurotani T, Komatsu Y, Kawanokuchi J, Kato H, Mitsuma N, Suzumura A. Neuroprotective role of phosphodiesterase inhibitor ibudilast on neuronal cell death induced by activated microglia. Neuropharmacology. 2004 Mar;46(3):404-11.
- Yoshioka M, Suda N, Mori K, Ueno K, Itoh Y, Togashi H, Matsumoto M. Effects of ibudilast on hippocampal long-term potentiation and passive avoidance responses in rats with transient cerebral ischemia. Pharmacological Research. 2002 Apr;45(4):305-11.
- Wakita H, Tomimoto H, Akiguchi I, Lin JX, Ihara M, Ohtani R, Shibata M. Ibudilast, a phosphodiesterase inhibitor, protects against white matter damage under chronic cerebral hypoperfusion in the rat. Brain Research. 2003 Nov 28;992(1):53-9.
- Rile G, Yatomi Y, Qi R, Satoh K, Ozaki Y. Potentiation of ibudilast inhibition of platelet aggregation in the presence of endothelial cells. Thrombosis Research. 2001 May 1;102(3):239-46.
- Feng J, Misu T, Fujihara K, Sakoda S, Nakatsuji Y, Fukaura H, Kikuchi S, Tashiro K, Suzumura A, Ishii N, Sugamura K, Nakashima I, Itoyama Y. Ibudilast, a nonselective phosphodiesterase inhibitor, regulates Th1/Th2 balance and NKT cell subset in multiple sclerosis. Multiple Sclerosis. 2004 Oct;10(5):494-8.
- Ledeboer A, Hutchinson MR, Watkins LR, Johnson KW. Ibudilast (AV-411). A new class therapeutic candidate for neuropathic pain and opioid withdrawal syndromes.Expert Opinion on Investigational Drugs. 2007 Jul;16(7):935-50.
- http://www.huffingtonpost.com/2013/04/03/meth-addiction-cure-ucla-ibudilast_n_2863126.html?utm_hp_ref=mostpopular#slide=more268305
Literature References:
Leukotriene D4 antagonist. Prepn: T. Irikura et al., DE 2315801; eidem, US 3850941 (1973, 1974 both to Kyorin).
Pharmacology and antiallergic activity: K. Nishino et al., Jpn. J. Pharmacol. 33, 267 (1983); H. Nagai et al., ibid. 1215.
In vitro cerebral vasodilating activity: M. Ohashi et al., Arch. Int. Pharmacodyn. 280, 216 (1986);
in vivo activity: W. M. Armstead et al., J. Pharmacol. Exp. Ther. 244, 138 (1988).
Bronchodilating activity in animals: S. Mue et al., Arch. Int. Pharmacodyn. 283,153 (1986).
Antiplatelet activity in animals: M. Ohashi et al., ibid. 321; M. Ohashi et al., Gen. Pharmacol.17, 385 (1986).

| PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|---|---|---|---|
| US4097483 | Aug 31, 1976 | Jun 27, 1978 | Kyorin Pharmaceutical Co., Ltd. | Pyrazolo 1,5-a!pyridines |
| US7585875 * | Jun 6, 2007 | Sep 8, 2009 | Avigen, Inc. | Phosphodiesterase inhibitors; neuropathic pain, inflammation, opioid dependence or withdrawal; 2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one for example |
| US20070015924 | Jun 15, 2006 | Jan 18, 2007 | Cardiome Pharma Corp. | stereoselective preparation of aminocyclohexyl ether compounds such as trans-(1R,2R)-aminocyclohexyl ether compounds and/or trans-(1S,2S)-aminocyclohexyl ether compounds; useful in treating arrhythmias |
| US20080070912 | Jun 6, 2007 | Mar 20, 2008 | Avigen, Inc. | Phosphodiesterase inhibitors; neuropathic pain, inflammation, opioid dependence or withdrawal; 2-amino-1-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)propan-1-one for example |
| US20090062330 | Jul 8, 2008 | Mar 5, 2009 | Medicinova, Inc. | Treatment of progressive neurodegenerative disease with ibudilast |
| US20090318437 * | Jun 10, 2009 | Dec 24, 2009 | Gaeta Federico C A | SUBSTITUTED PYRAZOLO[1,5-a] PYRIDINE COMPOUNDS AND THEIR METHODS OF USE |
| WO2007142924A1 | May 29, 2007 | Dec 13, 2007 | Avigen Inc | Ibudilast for inhibiting macrophage migration inhibitory factor (mif) activity |
| WO2007146087A2* | Jun 6, 2007 | Dec 21, 2007 | Avigen Inc | SUBSTITUTED PYRAZOLO [1,5-α] PYRIDINE COMPOUNDS AND THEIR METHODS OF USE |
| WO2010151551A1* | Jun 22, 2010 | Dec 29, 2010 | Medicinova, Inc. | ENANTIOMERIC COMPOSITIONS OF 2-AMINO-1-(2-ISOPROPYLPYRAZOLO[1,5-a]PYRIDIN-3-YL)PROPAN-1-ONE AND RELATED METHODS |
| US8119657 | Jun 22, 2010 | Feb 21, 2012 | Medicinova, Inc. | Enantiomeric compositions of 2-amino-1-(2-isopropylpyrazolo[1,5-α]pyridin-3-yl)propan-1-one and related methods |
| PATENT | FILING DATE | PUBLICATION DATE | APPLICANT | TITLE |
|---|---|---|---|---|
| WO2003104178A1* | Jun 6, 2003 | Dec 18, 2003 | Cortical Pty Ltd | Napththalene derivatives which inhibit the cytokine or biological activity of macrophage migration inhibitory factor (mif) |
| WO2003104203A1* | Jun 6, 2003 | Dec 18, 2003 | Cortical Pty Ltd | Therapeutic molecules and methods-1 |
| WO2004058713A1* | Dec 18, 2003 | Jul 15, 2004 | Jason Chyba | Differential tumor cytotoxocity compounds and compositions |
| WO2005058304A1* | Dec 17, 2004 | Jun 30, 2005 | Cortical Pty Ltd | Implantable device containing inhibitor of macrophage migration inhibitory factor |
| WO2006045505A1* | Oct 19, 2005 | May 4, 2006 | Novartis Ag | Mif-inhibitors |
| WO2006108671A1* | Apr 13, 2006 | Oct 19, 2006 | Novartis Ag | 3,4-dihydro-benzo[e][1,3]oxazin-2-ones |
Keywords: Antiallergic; Antiasthmatic (Nonbronchodilator); Leukotriene Antagonist; Vasodilator (Cerebral).

No comments:
Post a Comment