 Chemists from The
Scripps Research Institute illustrate their powerful new technique to
make and modify medicines, published recently in the journal Science.
Professor Phil Baran (center) holds a flask representing amines to
cleave strained C–C bonds, which are depicted by a tug-of-war between
co-first authors Ryan Gianatassio (left center) and Justin Lopchuk
(right center) with co-authors Chung-Mao Pan (left) and Jie Wang.
Chemists from The
Scripps Research Institute illustrate their powerful new technique to
make and modify medicines, published recently in the journal Science.
Professor Phil Baran (center) holds a flask representing amines to
cleave strained C–C bonds, which are depicted by a tug-of-war between
co-first authors Ryan Gianatassio (left center) and Justin Lopchuk
(right center) with co-authors Chung-Mao Pan (left) and Jie Wang.

Bicyclo[1.1.1]pentan-1-amine
Bicyclo[1.1.1]pentan-1-amine hydrochloride
http://www.scripps.edu/news/press/2016/20160114baran.html
TSRI Chemists Devise Powerful New Method for Modifying Drug Molecules
‘Strain-release amination’ technique emerged from efforts to help Pfizer synthesize promising cancer drug candidate
LA JOLLA, CA—January 14, 2016—Chemists at The Scripps Research
Institute (TSRI) have developed a versatile new technique for making
modifications—especially one type of extremely difficult, but
much-sought-after modification—to complex drug molecules.
The feat, reported in the January 15 issue of the journal
Science,
has already enabled pharma giant Pfizer to proceed with the evaluation
of a promising cancer drug candidate that otherwise could not have been
made in sufficient quantities.
“People from other pharma companies who have seen early drafts of
this paper can’t get their hands on the supporting information fast
enough,” said senior investigator Phil S. Baran, the Darlene Shiley
Professor of Chemistry at TSRI. “I expect that every company in the
business of making drugs will be using this chemistry soon.”
The technique, known as “strain-release amination,” also should
enable the easier construction of a variety of molecules besides
pharmaceuticals, including molecular probes for basic biology studies,
plastics, and other materials made from organic compounds.
Pfizer’s Bottleneck
The project began with Pfizer’s request for help in synthesizing a
molecule known as bicyclo[1.1.1]pentan-1-amine, which it needed to make
the cancer drug candidate. The Baran laboratory frequently collaborates
with Pfizer and other pharma companies to solve tough problems in
medicinal and process chemistry.
Traditional methods of synthesizing bicyclo[1.1.1]pentan-1-amine left
much to be desired. “Most of the previously published synthetic routes
require three to five steps with toxic reagents and yield only tens of
milligrams,” said Ryan Gianatassio, a PhD student at TSRI who was
co-first author of the study.
Pfizer needed kilograms of bicyclo[1.1.1]pentan-1-amine for
preclinical studies of its cancer drug candidate, and the company had
had to shelve the drug’s development until it could make that much of
it.
“We built a team of expert synthetic chemists to solve this
challenging problem, including chemists from Phil Baran’s lab and
Pfizer’s synthetic and process chemistry groups,” said Michael R.
Collins, a senior principal scientist at the drug company’s La Jolla
Laboratories.
Baran and his team, including Gianatassio and co-first author TSRI
Research Associate Justin M. Lopchuk, were able to solve the supply
problem for this building block, enabling a relatively quick and easy
synthesis from a readily available starting compound. “Using our
procedure, Pfizer easily produced over 100 grams, and they are now in a
position to scale that up further and re-start that delayed drug
development program,” said Gianatassio.
Adding Strained-Ring Structures
Baran realized that the new method could have much broader applications.
Bicyclo[1.1.1]pentan-1-amine is a “spring-loaded” or “strained ring”
molecule, in which carbon atoms are arranged in rings at odd angles,
with relatively large bond energies. Pharmaceutical chemists know that
adding such a structure to a drug molecule sometimes greatly improves
the drug’s properties: making it more absorbable by the gut, for
example, or enabling it to resist breakdown by enzymes in the body so
that it works therapeutically for longer periods.
The problem has been that, using traditional methods, the insertion
of these small structures into larger drug molecules is tricky—so much
so that chemists often have had to redesign the entire synthesis around
the small added structure.
“The way they’ve been doing it is like decorating a Christmas tree by
putting the ornaments in place first and then growing the tree around
it,” said Baran. “In many cases they just won’t pursue that because of
the time and labor it would take.”
Baran and his team showed that they could use their new method to
directly append a strained-ring molecule favored by pharmaceutical
chemists—propellane, so-called because its structure resembles a
propeller—to existing larger drug molecules. “We can make that
five-carbon ring structure of propellane click onto a wide range of drug
molecules of a type known as secondary amines—we call that a
propellerization reaction,” said Lopchuck.
“In fact, starting with a stock solution of the propellane, we can
use high-throughput techniques to quickly elaborate a matrix of
amine-containing compounds with the bicyclopentyl moiety, instead of
painstakingly synthesizing the compounds one at a time,” Collins said.
The team went on to demonstrate similar direct modifications using
two other strained-ring structures, azetidine and cyclobutane.
The TSRI researchers also found that they could use the new method to
attach molecules very precisely and selectively to specific amino acids
on proteins, thus in principle enabling the creation of new biologic
drugs as well as new reagents that would be useful in basic biology
research. “This technique opens up a world of chemistry that academic
and commercial laboratories have really wanted to look into but
couldn’t, due to the technical obstacles,” said Baran.
The supporting, publicly available information on strain-release
amination is meant to enable chemists to start using the technique right
away. A behind-the-scenes account and high-definition photos of the new
reaction setup can be found on the Baran Lab Blog,
Open Flask.
“This can be considered rapid bench-to-bedside chemistry because it
is fundamental science that will have a positive impact on human
medicine in a short period of time,” Baran said.
Other co-authors of the paper, “Strain Release Amination,” were Jie
Wang, Chung-Mao Pan, Lara R. Malins and Liher Prieto of TSRI; and Thomas
A. Brandt, Gary M. Gallego, Neal W. Sach, Jillian E. Spangler, Huichun
Zhu and Jinjiang Zhu, of Pfizer.
The research was funded in part by Pfizer and the National Institutes
of Health’s National Institute of General Medical Sciences.
About The Scripps Research Institute
The Scripps Research Institute (TSRI) is one of the
world's largest independent, not-for-profit organizations focusing on
research in the biomedical sciences. TSRI is internationally recognized
for its contributions to science and health, including its role in
laying the foundation for new treatments for cancer, rheumatoid
arthritis, hemophilia, and other diseases. An institution that evolved
from the Scripps Metabolic Clinic founded by philanthropist Ellen
Browning Scripps in 1924, the institute now employs about 2,700 people
on its campuses in La Jolla, CA, and Jupiter, FL, where its renowned
scientists—including two Nobel laureates—work toward their next
discoveries. The institute's graduate program, which awards PhD degrees
in biology and chemistry, ranks among the top ten of its kind in the
nation. For more information, see
www.scripps.edu.
# # #
For information:
Office of Communications
Tel: 858-784-2666
Fax: 858-784-8136
press@scripps.edu

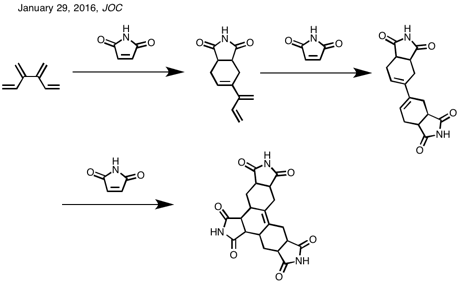
Org Lett. 2014 Apr 4;16(7):1884-7. doi: 10.1021/ol500635p. Epub 2014 Mar 14.
A new route to bicyclo[1.1.1]pentan-1-amine from 1-azido-3-iodobicyclo[1.1.1]pentane.
Abstract
From
a medicinal chemistry perspective, bicyclo[1.1.1]pentan-1-amine (1) has
served as a unique and important moiety. Synthetically, however, this
compound has received little attention, and only one scalable route to
this amine has been demonstrated. Reduction of an easily available and
potentially versatile intermediate, 1-azido-3-iodobicyclo[1.1.1]pentane
(2), can offer both a flexible and scalable alternative to this target.
Herein, we describe our scrutiny of this reportedly elusive
transformation and report our ensuing success with this endeavor.

Scalable Synthesis of 1-Bicyclo[1.1.1]pentylamine via a Hydrohydrazination Reaction
Pfizer
Worldwide Research and Development, La Jolla Laboratories, 10770
Science Center Drive, San Diego, California 92121, United States
Org. Lett., 2011, 13 (17), pp 4746–4748
DOI: 10.1021/ol201883z
Publication Date (Web): August 11, 2011
Copyright © 2011 American Chemical Society
Abstract
The reaction of [1.1.1]propellane with di-tert-butyl azodicarboxylate and phenylsilane in the presence of Mn(dpm)3 to give di-tert-butyl 1-(bicyclo[1.1.1]pentan-1-yl)hydrazine-1,2-dicarboxylate is described. Subsequent deprotection gives 1-bicyclo[1.1.1]pentylhydrazine followed by reduction to give 1-bicyclo[1.1.1]pentylamine. The reported route marks a significant improvement over the previous syntheses of 1-bicyclo[1.1.1]pentylamine in terms of scalability, yield, safety, and cost.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO .....FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook
 FACEBOOK
FACEBOOK
Join me on twitter

 amcrasto@gmail.com
amcrasto@gmail.com
/////









 .
.




 .
.

